
Synthesis and structure of novel Ag2Ga2SiSe6 crystals: promising materials for dynamic holographic image recording

Abstract
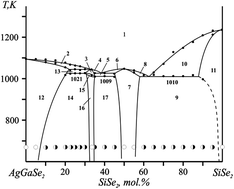
Phase diagrams of the AgGaSe2–SiSe2 system were explored by differential thermal analysis (DTA) and X-ray diffraction (XRD) analysis methods for the first time. It was demonstrated that the investigated system forms quaternary compounds of compositions Ag2Ga2SiSe6 and AgGaSiSe4. Ag2Ga2SiSe6 melts at 1042 K and exists in two polymorphous modifications. The crystal structure of the low-temperature modification was determined by the single crystal method (space group I![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) 2d (122) and lattice parameters a = 5.9021(1) Å, b = 5.9021(1) Å, and c = 10.4112(10) Å). Additional details (CIF file) regarding the crystal structure investigations are available at the Fachinformationszentrum Karlsruhe. The band gap (Eg) of the Ag2Ga2SiSe6 system was estimated from the fundamental absorption edge and we showed that it decreases with increasing temperature (100–300 K) from 2.13 eV to 1.97 eV. The compound is photosensitive and its spectral dependence on the photoconductivity has two maxima: at λmax1 = 640 nm and λmax2 = 900 nm. For the pristine Ag2Ga2SiSe6 crystal surface, X-ray photoelectron core-level and valence-band spectra were obtained. The X-ray photoelectron valence-band spectrum of Ag2Ga2SiSe6 was compared on a common energy scale with the X-ray emission Se Kβ2 and Ga Kβ2 bands, representing peculiarities of the energy distribution of the Se 4p and Ga 4p states, respectively. The comparison revealed that the principal contributions of the valence Se p and Ga p states occur in the upper and central parts of the valence band, respectively, with significant contributions to other valence band regions. The illumination by the bicolour coherent pulses of the Er:glass nanosecond lasers at different angles led to the formation of the gratings, which are sensitive to the irradiation time.
2d (122) and lattice parameters a = 5.9021(1) Å, b = 5.9021(1) Å, and c = 10.4112(10) Å). Additional details (CIF file) regarding the crystal structure investigations are available at the Fachinformationszentrum Karlsruhe. The band gap (Eg) of the Ag2Ga2SiSe6 system was estimated from the fundamental absorption edge and we showed that it decreases with increasing temperature (100–300 K) from 2.13 eV to 1.97 eV. The compound is photosensitive and its spectral dependence on the photoconductivity has two maxima: at λmax1 = 640 nm and λmax2 = 900 nm. For the pristine Ag2Ga2SiSe6 crystal surface, X-ray photoelectron core-level and valence-band spectra were obtained. The X-ray photoelectron valence-band spectrum of Ag2Ga2SiSe6 was compared on a common energy scale with the X-ray emission Se Kβ2 and Ga Kβ2 bands, representing peculiarities of the energy distribution of the Se 4p and Ga 4p states, respectively. The comparison revealed that the principal contributions of the valence Se p and Ga p states occur in the upper and central parts of the valence band, respectively, with significant contributions to other valence band regions. The illumination by the bicolour coherent pulses of the Er:glass nanosecond lasers at different angles led to the formation of the gratings, which are sensitive to the irradiation time.
 Please wait while we load your content...
Please wait while we load your content...

