
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Aptamer mediated niosomal drug delivery†
Didem Ag
Seleci
,
Muharrem
Seleci
,
André
Jochums
,
Johanna-Gabriela
Walter
,
Frank
Stahl
* and
Thomas
Scheper
Institute of Technical Chemistry, Leibniz University Hannover, Callinstreet 5, 30167 Hannover, Germany. E-mail: stahl@iftc.uni-hannover.de
First published on 6th September 2016
Abstract
Development of nanoscale carrier systems for targeted drug delivery is crucial for cancer treatment. The current methods of drug delivery exhibit some problems such as lack of therapy efficiency at the desired parts of the body, degradation of the drug before reaching the desired tissue and limitations in cellular penetration. In this work, a novel drug delivery platform was developed to overcome these problems and to enable specific and efficient uptake into the cells. The surface of the synthesized polyethylene glycolated niosomes (PEGNIO) was modified with cell penetrating peptide (CPP) and cell specific MUC1 (S2.2) aptamer, and doxorubicin (DOX) as a cancer model drug was encapsulated in this platform. Fluorescence microscopy and flow cytometry analysis were used to investigate the cellular uptake and intracellular distribution of the DOX loaded niosomal formulation. In vitro cytotoxicity studies were carried out using MUC1 positive HeLa and negative U87 cells. Moreover, dynamic light scattering (DLS), zeta potential measurements and fluorescence absorption spectroscopy were performed to determine the vesicle size, as well as charge and spectroscopic properties of the conjugates. From these results, this novel aptamer mediated niosomal drug delivery platform may have application potential in targeted drug delivery towards MUC1-overexpressing tumors.
Introduction
A drug delivery system (DDS) is described as a formulation that enables the introduction of drug molecules into the body with improved efficacy and safety. Due to the rapid progress of nanotechnology, numerous nanocarriers have been developed to securely deliver drugs into target sites. New targeting agents, including aptamers, short peptides, and small molecules have recently become promising targeting ligands to design novel drug delivery systems.The current treatment options for cancer are surgical operation, radiation and chemotherapy or a combination. The therapeutic efficacy of many anticancer drugs is limited by their poor penetration into tumor tissue and by their side effects on healthy cells.1 To overcome these limitations, development of a novel carrier platform for specific drug uptake into the cell with an optimal dose at high efficiency is important. Therefore, anticancer drugs can be conjugated with biomolecules exhibiting potential for cellular targeting and penetration and can be delivered to the desired site of action by multi-functional carrier platforms. Vesicular nanocarriers have received great attention as potential drug carrier systems. Nonionic surfactant based vesicles “niosomes” are one class of vesicular nanocarriers which can accommodate a large number of drugs with a wide range of solubility.2–4 Due to their low cost, long term storage stabilities and lower toxicity of niosomes have been used for drug delivery.5–7 The addition of polyethylene glycol (PEG) to the nanocarrier surface increases the steric stabilization of the nanoparticle and allows for further surface modifications to design specific drug delivery systems. It is generally thought that PEGylation protects the delivery systems against the immune system and thereby prolong circulation life times.8
In solid tumors, the penetration of the tissue by the anticancer drug is limited which causes reduced efficacy and the development of drug resistance.9 A promising approach to overcome the cellular barrier is based on the use of certain peptides namely cell penetrating peptides (CPPs), able to translocate across the cell membrane and deliver their payload intracellularly within minutes.10 CPPs consist of small cationic or amphipathic peptides that aid the uptake of attached cargos into living cells. A wide variety of small molecules and biomolecules including plasmid DNA, siRNA, oligonucleotides and peptide nucleic acid molecules have been attached to these peptides and were subsequently internalized.11–13 The ability of CPPs to translocate biologically active molecules into cells makes these peptides promising candidates for theranostic applications.14 TAT is one of the smallest polycationic CPPs composed of arginine and lysine residues. Studies on the binding affinities of cationic TAT peptides indicate that these peptides strongly bind electrostatically to the various anionic species (e.g. heparan sulphate proteoglycans) present at the extracellular surface of cell membranes.15,16 The exact molecular mechanism of cellular entry of CPPs is currently not fully understood. Former studies indicated that in general uptake occurs by endocytosis (or more specifically macropinocytosis) and direct membrane translocation.17 The lack of cell specificity remains the major drawback for the clinical application of CPPs.18
Receptors that are over-expressed in many cancer cells are suitable targets to achieve a more specific delivery. MUC1 is a large transmembrane glycoprotein overexpressed in most malignant adenocarcinoma, including ovarian, lung, pancreatic, prostate, and breast cancers, making it an ideal target molecule for chemotherapeutics.19 Aptamers are short oligonucleotides that are capable to selectively bind their corresponding target.20–22 These reagents are selected by an in vitro process called SELEX, (systematic evolution of ligands by exponential enrichment).23 Several MUC1 aptamers were developed by Ferreira et al. and S2.2 is a 25-nucleotide truncated version of the original MUC1 aptamer. It binds MUC1 protein with high specificity and affinity with a KD of 0.135 nM.24,25 S2.2 has been used in a few targeted delivery systems. Yu et al. used MUC1 aptamer to target paclitaxel (PTX) loaded poly(lactic-co-glycolic-acid) (PLGA) nanoparticles.26 Furthermore PEG-modified MUC1 targeting doxorubicin (DOX) was designed and the aptamer–doxorubicin complex was prepared by intercalation of the aptamer with DOX by Tan et al.27 Recently, Liu et al. synthesized vinorelbine (VRL) loaded and MUC1 aptamer modified lipid–polymer hybrid nanoparticles.22
In this study, polyethylene glycolated niosomes (PEGNIO) were prepared from span60, cholesterol and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[maleimide (polyethylene glycol)-2000] (DSPE-PEG(2000) Maleimide). DOX was encapsulated into the PEGNIO. The niosomes were characterized with respect to size, morphology and drug encapsulation efficiency. Cysteine-modified cell penetrating peptide (CysTAT) was conjugated to the amine group of MUC1 aptamer in the presence of a crosslinking agent bis(sulfosuccinimidyl)suberate (BS3). Subsequently CysTAT–MUC1 conjugate was attached to DOX encapsulated PEGNIO (PEGNIO/DOX) via the formation of a thioether linkage. The anticancer activity of DOX-loaded targeted vesicles was studied in HeLa (cervical cancer cells) and U87 (human glioblastoma cells) cell lines by evaluating the cellular uptake and cytotoxicity. The designed nanoparticular drug delivery system composed of aptamer–CPP–PEGNIO was first fabricated in this study. Our findings suggest that this platform can serve as a delivery vehicle for cancer cells overexpressing MUC1.
Materials and methods
Materials
Amine and Cy5 modified MUC1 aptamer S2.2 (5′-NH2-GCA GTT GAT CCT TTG GAT ACC CTG G-3′), (5′-Cy5-GCA GTT GAT CCT TTG GAT ACC CTG G-3′) were purchased from Integrated DNA Technologies. CysTAT with CYGRKKRRQRRR–NH2 sequence was obtained from GenScript. DSPE-PEG(2000) Maleimide was provided by Avanti. Span60, cholesterol, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazoliumbromide (MTT), 4,6-diamino-2-phenylindol (DAPI) and Dulbecco's Modified Eagle Medium (DMEM) were ordered from Sigma Aldrich. RNAtidy G and sodium dodecyl sulfate (SDS) were purchased from Applichem. BS3 was ordered from Covachem. Doxorobucin (hydrochloride) was purchased from Cayman Chemical. GeneRuler 100 bp DNA ladder and dNTP Set (100 mM solutions) were obtained from Fermentas. M-MLV reverse transcriptase and its M-MLV RT 5× buffer as well as GoTaq polymerase and its 5× Green GoTaq reaction buffer were provided from Promega. PCR primers were synthesized by Life Technologies.Preparation of PEGylated niosomes
Multi lamellar vesicles (MLVs) of PEGNIO were prepared by the thin film hydration method.28 Span 60, cholesterol, and DSPE-PEG(2000) Maleimide were dissolved in 1.0 mL chloroform in a round-bottom flask with the molar ratio of 4.95 mM![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4.95 mM:0.1 mM. The solvent was evaporated with constant rotation at 38 °C under reduced pressure to form a thin lipid film. Then the thin film was hydrated with 1.0 mL of distilled water for preparing empty niosomes, or a doxorubicin solution (0.22 × 10−3 M in water) to obtain PEGNIO/DOX conjugate at 60 °C for 60 min. Afterward the niosomal suspension was equilibrated at room temperature overnight, to complete annealing and partitioning of the drug between the lipid bilayer and the aqueous phase.5 Small unilamellar vesicles (SUVs) were prepared starting from MLVs by extruding the niosomes 11 times through 0.4 μm and 0.1 μm pore size polycarbonate filters using mini-extruder set (Avanti polar lipids). Niosomes were purified by dialysis against water for 3 h using 6–8 kDa dialysis bag.
:4.95 mM:0.1 mM. The solvent was evaporated with constant rotation at 38 °C under reduced pressure to form a thin lipid film. Then the thin film was hydrated with 1.0 mL of distilled water for preparing empty niosomes, or a doxorubicin solution (0.22 × 10−3 M in water) to obtain PEGNIO/DOX conjugate at 60 °C for 60 min. Afterward the niosomal suspension was equilibrated at room temperature overnight, to complete annealing and partitioning of the drug between the lipid bilayer and the aqueous phase.5 Small unilamellar vesicles (SUVs) were prepared starting from MLVs by extruding the niosomes 11 times through 0.4 μm and 0.1 μm pore size polycarbonate filters using mini-extruder set (Avanti polar lipids). Niosomes were purified by dialysis against water for 3 h using 6–8 kDa dialysis bag.
Synthesis and characterization of CysTAT–MUC1 conjugate
The conjugation between CysTAT and amine modified MUC1 aptamer was performed using an amine to amine crosslinker BS3. 35 μL containing 5 nmol amine modified MUC1 aptamer, 30 μL containing 20 nmol CysTAT peptide and 150 μL containing 1.75 μmol BS3 were mixed in 85 μL 0.1 M sodium phosphate including 0.15 M sodium chloride at pH 7.4 and incubated at room temperature for 1 h. Once the reaction was completed, 300 μL 1.0 M Tris buffer was added to quench the reaction for 20 minutes at room temperature. Unreacted peptides and quenched crosslinkers were removed through dialysis against PBS (pH 7.2) using 3.5 kDa dialysis bag. During this reaction aptamer–aptamer and peptide–peptide conjugation can occur. Denaturing urea polyacrylamide gel electrophoresis (Urea PAGE) and HPLC were used for the characterization of CysTAT–MUC1. Urea PAGE was performed according to manufacturer's instructions (QIAGEN). Briefly 15% acrylamide/urea gel was prepared and run for 30 min at 200 V. The samples were heated at 95 °C for 2 min and then they were immediately transferred onto ice. The samples were loaded onto the gel and the gel was run for 1.5 h at 200 V. Afterward the gel was stained with methylene blue solution and documented using an INTAS UV documentation system. HPLC measurements and carried out using VWR Hitachi Chromaster. For the analysis, a DAD detector and Kinetix 2.6 μm C8 100 Å, 150 × 4.6 mm (Phenomenex) column were used. Detection was performed at 214 nm at room temperature. The mobile phase consisted of 0.065% trifluoroacetic acid (TFA) in water and 0.05% TFA in acetonitrile with a flow rate of 1.0 mL min−1.Conjugation CysTAT–MUC1 to PEGNIO/DOX
CysTAT–MUC1 was conjugated to PEGNIO/DOX via thiol group of cysteine to maleimide group on PEGNIO resulting in the formation of a thioether linkage.29,30 Maleimide group reacts specifically with thiol-groups in the pH range 6.5–7.5. 100% of the prepared CysTAT–MUC1 conjugate in PBS (pH 7.2) and 50% of the prepared PEGNIO/DOX were mixed and incubated overnight at room temperature. After completing the reaction between sulfhydryl group of CysTAT and maleimide group of DSPE-PEG(2000) Maleimide, the final PEGNIO/DOX/CysTAT–MUC1 conjugate was purified using 14 kDa dialysis bag to remove unbound CysTAT–MUC1. Schematic representation of niosome synthesis, drug encapsulation and the bioconjugation processes are shown in Scheme 1.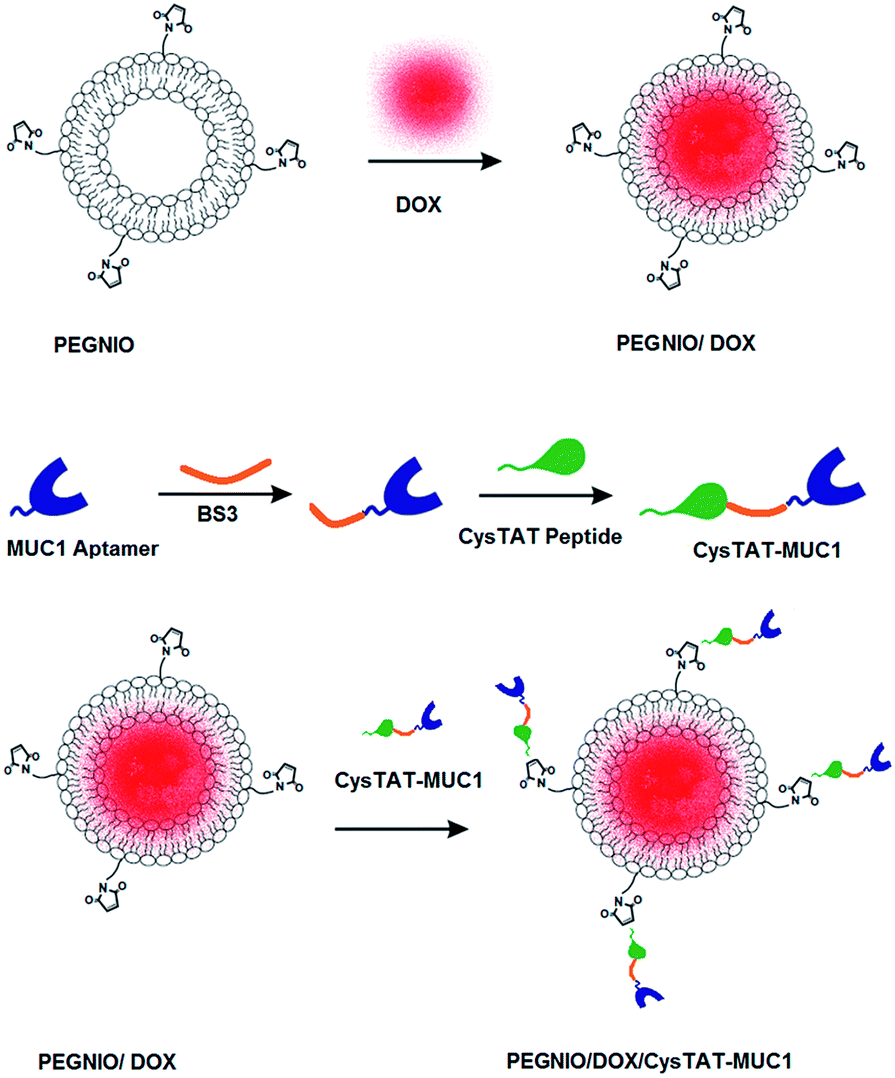 | ||
| Scheme 1 Schematic representation of drug the encapsulation and the bioconjugation process. | ||
Measurement of particle size, distribution and zeta potential
Size, size distribution and zeta potential of niosomes were determined by dynamic light scattering (DLS) analysis using Malvern Zetasizer Nanoseries-Nano-ZS. The polydispersity index (PDI) was used as a measure of the width of size distribution. PDI less than 0.3 corresponds to a homogenous population for colloidal systems.31 Each sample was measured three times.Stability
The stability of DOX loaded niosomal formulations was tested via DLS analysis. After the synthesis of PEGNIO/DOX and PEGNIO/DOX/CysTAT–MUC1, these conjugates were stored at 4 °C in the dark. The particle size and PDI values were measured for 2 months. Additionally the particle size of PEGNIO/DOX/CysTAT–MUC1 was measured in cell culture media before and after the incubation at 37 °C for 24 h.Entrapment efficiency
The drug encapsulation efficiency was determined using the dialysis technique.32 According to this method, directly after the preparation 1.0 mL of PEGNIO/DOX and 1.0 mL of PEGNIO/DOX/CysTAT–MUC1 dispersions were dropped into two dialysis bags (12–14 kDa) and immersed in 100 mL of distilled water with magnetic stirring at 100 rpm. Samples were dialyzed for 3 h. The percent of encapsulation efficiency (E%) was expressed as the percentage of the drug entrapped in niosomes (and thus not removed via dialysis) referred to the initial amount of drug that is present in the nondialyzed sample. It was determined by diluting 50 μL of dialyzed and 50 μL of nondialyzed niosomes in 1.0 mL of methanol. This step is essential for breaking the niosomal membrane, thereby releasing the entrapped DOX. Subsequently, the amount of DOX was determined by HPLC using a C18 column (Phenomenex Kinetix, 4.6 × 100 mm, 2.6 μm) at 35 °C and a UV detector was conducted at 254 nm. The mobile phase consisted of methanol and water (60/40, v/v) containing 0.1% formic acid and 0.1% ammonia solution (25%) with a flow rate of 1.0 mL min−1.33 The stock solutions of DOX were prepared at 1.0 mg mL−1 in methanol and further diluted with methanol in the concentration range 1.0–200 μg mL−1. The amount of encapsulated DOX was calculated according to the calibration curve (y = 120777x − 67040, R2 = 0.9984). The limit of detection (LOD) and limit of quantification (LOQ) for DOX were found to be 6.12 μg mL−1, 18.55 μg mL−1 respectively based on 3.3σ/slope and 10σ/slope formulations.34
Drug release
Drug release experiments were performed using the dialysis method. The DOX-loaded niosome solutions were prepared and transferred into a dialysis membrane tubing (Thermo, Slide-ALyzer MINI Dialysis Devices, 10k MWCO). The tubing was immersed in 10 mL of the PBS buffer (pH 5.6 and 7.4), placed in an incubator at 37 °C and stirred at 100 rpm. At specific time intervals, 0.5 mL samples were removed from the release medium and replaced with the same volume of fresh buffer. A calibration curve was established with a known concentration of free DOX by fluorescence emission measurements at 595 nm using NanoDrop 3300. The amount of released DOX was calculated according to the calibration curve (y = 254.93x − 30.74, R2 = 0.9960, LOD = 0.61 μg mL−1, LOQ = 1.84 μg mL−1).Cell culture
HeLa and U87 cell lines were provided from German Collection of Microorganisms and Cell Cultures (DSMZ). Both cell lines were grown in DMEM containing 10% fetal calf serum (FCS) and 1.0% penicillin/streptomycin (P/S). All cells were cultivated in medium and incubated with samples and reagents at 37 °C in a humidified environment with 5.0% CO2.MUC1 expression on cell surfaces
PCR and flow cytometry analysis were used to confirm expression of the MUC1 receptor in HeLa and lack of expression in U87 cells. Total cellular RNA of the cells was isolated using Trizol reagent (Invitrogen) and transcribed into cDNA. The primers were designed with Lasergene Primer Select Software using the NCBI reference mRNA sequence for Homosapiens mucin 1, cell surface associated (MUC1), transcript variant 1 (NM_002456.5). The primer sequences are as follows: MUC1 forward 5′-TAC CGA TCG TAG CCC CTA TG-3′ and reverse 5′-CCA CAT GAG CTT CCA CAC AC-3′. The human housekeeping gene hypoxanthine phosphoribosyltransferase (HPRT) was additionally used to prove the successful synthesis of cDNA. The primer sequences are: HPRT forward 5′-AAG CTT GCT GGT GAA AAG GA-3′ and reverse 5′-AAG CAG ATG GCC ACA GAA CT-3′. The protocol described in our previous publication was used in the polymerase chain reactions.35 The annealing temperature of 60 °C was used for MUC1 and HPRT during PCR experiments. PCR products were separated in 1.5% agarose gel in TAE buffer and stained with 5.0 μL/100 mL buffer Roti-Safe Gel Stain ready to use by Thermo. The gel was run using the Thermo EC electrophoresis unit at 100 V for 60 min and documented using an INTAS UV documentation system.For flow cytometry studies, 5 × 105 cells were collected. 100 μL of 5.0 μM Cy5 labelled MUC1 aptamer in PBS were added to the cells and the cell suspension was shaken at room temperature for 1 h with 500 rpm in the dark. The cells were washed once in 300 μL of PBS to remove unbound aptamer. Before flow cytometric analysis, cells were resuspended in 500 μL of PBS and the stained cells were analyzed in a BD Accuri C6 flow cytometer. At least 20000 gated events were observed in total and living cells were gated in a dot plot of forward versus side scatter signals. For drawing dot plots and histograms the BD Accuri C6 software was used.
Cytotoxicity
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assays were used to determine cytotoxicity of the niosomal formulations. Cells (8 × 103) were seeded out in 96-well tissue plates (Sarstedt, USA) in a volume of 200 μL and cultivated for three days. After this cultivation time cells were washed once in PBS and treated with SUVs (PEGNIO, PEGNIO/DOX, PEGNIO/DOX/CysTAT–MUC1) and free DOX for 24 h and 48 h. The equivalent concentration of free DOX was used in niosomal formulations. Then the samples were removed and cells were incubated in 110 μL per well 10% MTT solution (5.0 mg mL−1 in PBS) in medium for 4 h. During this incubation time, formazan complex was produced by the cells. 100 μL SDS solution (1.0 g SDS in 10 mL 0.01 M HCl) was added to each well to release the purple colored salt from the cells. After 24 h of incubation, UV-vis absorption was measured at 570 nm to 630 nm as the reference wavelength using a microplate reader Epoch Biotek.Cellular uptake and internalization
The DOX uptake by HeLa and U87 cells for different DOX formulations was analyzed by flow cytometry. HeLa and U87 cells were treated with PEGNIO/DOX, PEGNIO/DOX/CysTAT–MUC1 and free DOX for 2 h and treated cells were washed two times with PBS, and then analyzed in a BD Accuri C6 flow cytometer.Cellular internalization of PEGNIO/DOX/CysTAT–MUC1 was determined via fluorescence microscopy studies. HeLa and U87 cells were cultivated for 2 days on the chamber slides (μ slides 8 well purchased at ibidi GmbH) in a volume of 200 μL of the medium. PEGNIO/DOX/CysTAT–MUC1 was diluted with medium and then added to the cells. The cells were incubated for 4 h at 37 °C and washed once in PBS. Afterward 100 μL DAPI solution (1.0 μg mL−1) was added to the cells and incubated for 15 min. After DAPI staining, the cells were washed with PBS once. Images were taken using an OLYMPUS BX41 fluorescence microscope equipped with an OLYMPUS SC30 camera.
Statistical analysis
Statistical analysis was performed using the GraphPad InStat statistical software. All experiments were repeated three times. The paired t-test was performed. The difference between two groups was considered to be significant when the p value was less than 0.05.Results
Doxorubicin is one of the most extensively used broad-spectrum anticancer drugs. It accumulates inside the cell nucleus where it intercalates into DNA and inhibits the progression of topoisomerase II to cause DNA damage and cleavage.36,37 Long treatment durations and toxic side effects are inconvenient in the use of conventional chemotherapeutics.38 Recent studies show that, biocompatible nanoparticles, with an increased surface area to volume ratio can overcome non-cellular and cellular-based mechanisms of resistance and increase the selectivity of drugs towards cancer cells, while reducing their toxicity towards normal tissues.39 The addition of PEG to the nanoparticle surface prolongs vesicles residence time in blood and accumulation at the pathological sites.5 Taking this into consideration, we decided to use PEGylated niosomes for obtaining aptamer targeted-cell penetrating vesicular systems.Conjugation and characterization of CysTAT–MUC1
BS3 is an amine reactive, homobifunctional, sulfo-NHS ester, crosslinking reagent.40 The reactivity of the sulfo-NHS esters is highly reactive toward amines in the pH range of 7–9. The amine modified MUC1 aptamer was reacted with the –NH2 group in the CysTAT peptide in the presence of BS3 to produce the CysTAT–MUC1 conjugates. Besides the desired conjugate CysTAT–MUC1, the reaction can also result in dimers of MUC1 aptamer and peptide. In order to investigate the product spectrum of the crosslinking reaction, gel electrophoresis was performed. Free MUC1 aptamer and CysTAT–MUC1 were applied to Urea PAGE. A single band was observed for free aptamer. In the case of CysTAT–MUC1 conjugate double bands were observed (Fig. S1†). In the double bands, first band indicates CysTAT–MUC1 conjugate and the second one exhibits unbound aptamers in the conjugate. Additionally, the conjugation of aptamer and peptide was proven by HPLC analysis. For this aim, chromatograms of MUC1 aptamer, BS3 crosslinker, Cys-TAT peptide were compared with chromatogram of CysTAT–MUC1 conjugate (Fig. S2†). No peaks were observed for MUC1 aptamer in this HPLC method. CysTAT peptide and BS3 crosslinker have different retention times 7.75 and 8.67 respectively, showed that there is no interference during the analysis of the CysTAT–MUC1 conjugates. The conjugation efficiency was about 82% in accordance with the integrated areas of CysTAT peptide before and after conjugation. Besides CysTAT–MUC1, the conjugation can also result in aptamer and peptide dimers. Nonetheless, since these side products are not able to bind to maleimide group of PEGNIO (due to the lack of –SH group), we used to the as prepared conjugate with no additional purification.Synthesis and characterizations of DOX loaded niosomal formulations
PEGylated niosomes were prepared by the thin film layer hydration technique, using span60:cholesterol:DSPE-PEG(2000) Maleimide. 0.22 mM doxorubicin solutions were used in the preparation of loaded vesicles. CysTAT–MUC1 was conjugated to PEGNIO/DOX to obtain targeted drug delivery system. The size of nanocarriers is very important for effective accumulation in tumor by enhanced permeability and retention effect (EPR) and cellular internalization. The mean diameters of empty and doxorubicin-loaded niosomal formulations, along with the corresponding polydispersity index (PDI), doxorubicin entrapment efficiency (E%) values, and zeta potential values are listed in Table 1. The empty vesicle size was analyzed to be 151.0 ± 36 nm. The hydrodynamic diameter did not change after DOX loading (152.7 ± 34 nm) but after conjugation with CysTAT–MUC1 it increased to 164.5 ± 40 nm. The stealth niosomes showed zeta potential values close to 0 mV but the surfaces of nanoparticles was grafted with PEG to improve water solubility and avoid aggregation.5,41,42 Due to the presence of PEG in the niosome structure, no aggregation was observed. DOX loading influenced the zeta potential of the vesicles. The change in the zeta potential may be a result of DOX intercalation in the vesicle membrane.5,43,44 Conjugation of CysTAT–MUC1 increased niosome size, corresponding with the presence of the aptamer on the niosomal surface increasing the hydrodynamic diameter. Moreover, the negatively charged DNA aptamer reduced the surface potential of the niosome.29 PDI ranged from 0.275 to 0.214, demonstrating that the vesicle population is relatively homogeneous in size. The DOX entrapment efficacy (E%) was calculated to be around 39% and 37% for PEGNIO/DOX and PEGNIO/DOX/CysTAT–MUC1 respectively. The stability of niosomal DOX formulations was tested via DLS analysis and no changes were observed in the size and PDI values after two months storage at 4 °C in the dark (data not shown). Additionally, PEGNIO/DOX/CysTAT–MUC1 sample was diluted in cell culture media and was incubated at 37 °C for 24 hours. The size of the sample was measured before and after incubation and no changes were observed.| Samples | Size (nm) intensity (%) (mean ± SD) | Polydispersity index (PDI) | Zeta potential (mV) | Entrapment efficiency (E%) |
|---|---|---|---|---|
| PEGNIO | 151.0 ± 36 | 0.244 | −4.96 ± 0.43 | — |
| PEGNIO/DOX | 152.7 ± 34 | 0.214 | −3.56 ± 0.27 | 39.52 ± 1.8 |
| PEGNIO/DOX/CysTAT–MUC1 | 164.5 ± 40 | 0.275 | −8.62 ± 0.50 | 37.48 ± 2.1 |
Drug release
Sustained drug release is one of the important properties of nanoscale drug delivery systems that will minimize side effects of the drug. In vesicular drug carrier systems, the drug release occurs by passive transport of the drug through the membrane bilayer.45 The release of DOX from PEGNIO/DOX/CysTAT–MUC1 was investigated using dialysis methods at pH 7.4, which was chosen in accordance with physiological conditions and in an acidic environment (pH 5.6). The solutions were taken out at specific intervals and measured by fluorescence emission measurements at 595 nm to determine the amount of DOX that has been released. The in vitro DOX release profiles from PEGNIO/DOX/CysTAT–MUC1 showed the faster release of DOX under acidic environments than that at neutral pH (Fig. 1). Within 48 h, the release of DOX was 30% and 52% at pH 7.4 and pH 5.6 respectively. This can be explained by the higher solubility of DOX at the lower pH.46 Especially, the pH change from 7.4 to 5.6 corresponds to the pH change from the normal physiological environment in body circulation to the intracellular tumor tissue. According to results this conjugate is expected to be a promising drug delivery system for the tumor-targeted delivery of DOX. | ||
| Fig. 1 In vitro cumulative release of DOX from PEGNIO/DOX/CysTAT–MUC1 at pH 7.4 and 5.6. | ||
Confirmation of MUC1 receptor expression on the cell surface
To test PEGNIO/DOX/CysTAT–MUC1 as a targeted drug delivery system in vitro, first the expression of MUC1 was evaluated in HeLa and U87 cells. Gene expression of MUC1 was investigated at the mRNA level using polymerase chain reaction (PCR) and at the cell surface protein level using flow cytometry. Agarose gel analysis with the results of PCR experiments is shown in Fig. 2. The housekeeping gen (HPRT) was measured as a control for both HeLa and U87 cells (Fig. 2a and b). Bands corresponding to housekeeping genes were observed for both cell types (263 bp) confirming the success of RNA extraction and PCR. Fig. 2a demonstrates that HeLa cells show a high level of transcription of the MUC1 gene, which results in a strong band at the expected base pair length of 283 bp. No corresponding band was observed in U87 cells, thereby indicating the lack of expression of MUC1. The PCR results were also confirmed by using Cy5 labeled MUC1 aptamer in flow cytometry. Cells were treated with a Cy5 labeled MUC1 aptamer. After the treatment, the mean fluorescence intensity was measured to be 28523 for U87 cells, and 57993 for HeLa cells (Fig. 2c). Both methods confirmed that MUC1 expression was considerably higher in HeLa cells than in U87 cells. These results are in agreement with the literature.47,48
 | ||
| Fig. 2 Image of 1.5% agarose gel with results of PCR for HPRT gene and MUC1 expression in HeLa and U87 cells (a and b). Flow cytometry analysis of MUC1 expression on HeLa and U87 cells using Cy5-labelled aptamer S2.2 (c). | ||
Cellular uptake and internalization
Flow cytometry was used to investigate the total DOX uptake by HeLa and U87 cells for different DOX formulations and to evaluate receptor mediated cell targeting. The cells were treated with samples for 2 h. Untreated control cells and treated cells were analyzed using a BD Accuri C6 flow cytometer. As shown in Fig. 3a, the cellular DOX level for PEGNIO/DOX/CysTAT–MUC1 in HeLa cells was higher than that of PEGNIO/DOX and free DOX. Free DOX enters the cells by diffusion, leading to higher drug levels than found with the PEGNIO/DOX. After encapsulation of DOX in PEGNIO, the DOX uptake by diffusion is reduced.49 In the case of U87 cells, the uptake of free DOX was higher than PEGNIO/DOX/CysTAT–MUC1 (Fig. 3b). Fig. 3c indicates that the synthesized PEGNIO/DOX/CysTAT–MUC1 conjugate bound to MUC1 positive HeLa cell specifically. Nonspecific binding to U87 cells was also observed, but the fluorescence signals were lower than for HeLa cells. Cellular internalization of PEGNIO/DOX/CysTAT–MUC1 obtained by fluorescence microscopy analysis. DOX is a fluorescent drug and, it localizes to the nucleus in tumor cells.50 Both cell lines were treated with PEGNIO/DOX/CysTAT–MUC1 for 4 h. The synthesized conjugate bound to HeLa cells, resulting in high fluorescence of the cell nucleus, thereby demonstrating successful internalization. In contrast, fluorescence was significantly lower for U87 cells, indicating some nonspecific uptake in U87 cells (Fig. 4a and b). Fluorescence microscopy images show results similar to flow cytometry analysis.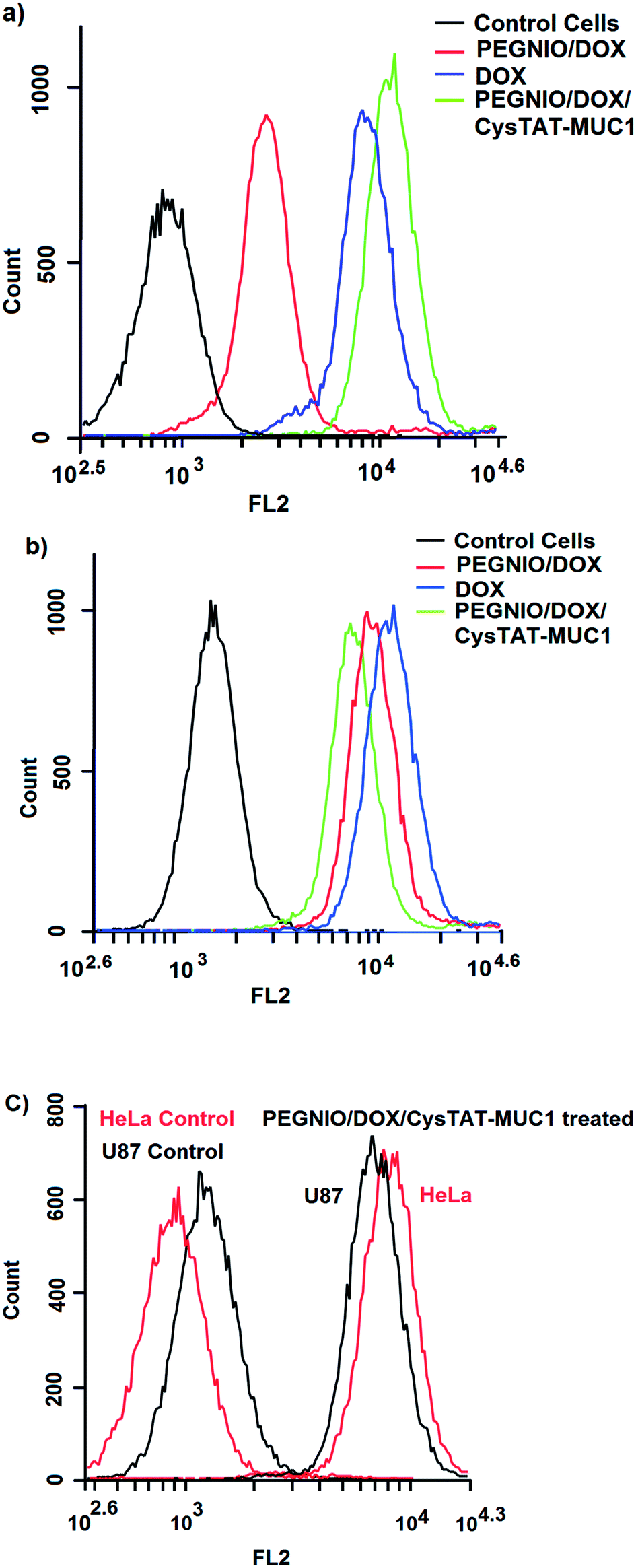 | ||
| Fig. 3 Flow cytometric measurement of DOX uptake by HeLa (a) and U87 cells (b) after incubating with PEGNIO/DOX, PEGNIO/DOX/CysTAT–MUC1 and free DOX. Histogram of binding of PEGNIO/DOX/CysTAT–MUC1 to MUC1 positive HeLa cells and MUC1 negative U87 cells (c). | ||
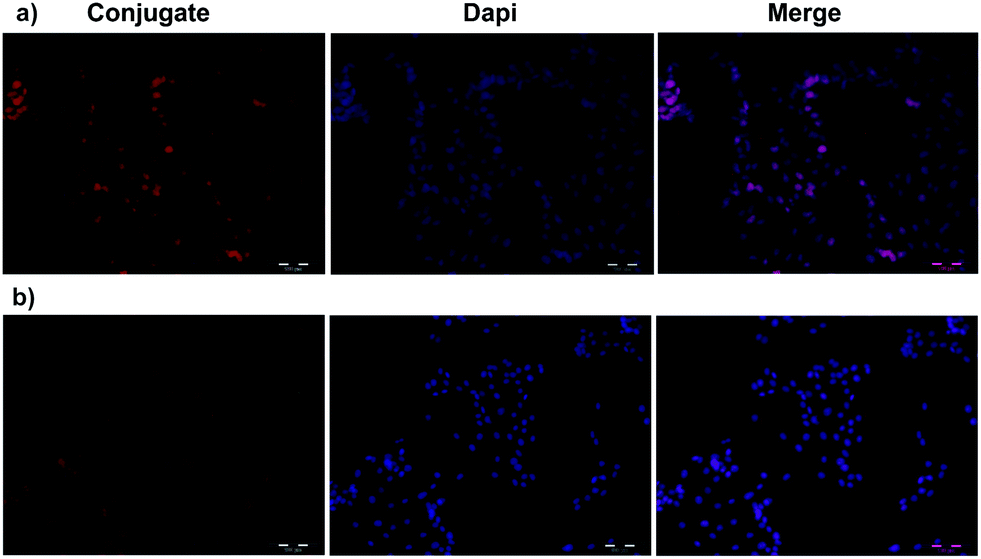 | ||
| Fig. 4 Fluorescence microscopy images of HeLa (a) and U87 cells (b). PEGNIO/DOX/CysTAT–MUC1 was incubated with the cells for 4 h at 37 °C. | ||
Cytotoxicity
The cytotoxicity of bare niosomes, drug loaded formulations and free DOX was investigated by MTT assay using HeLa and U87 cells. PEGNIO was practically nontoxic to HeLa and U87 cells with relative cell viabilities above 80% for both 24 and 48 h (Fig. 5). PEGNIO/DOX was less toxic than free DOX on both cell lines after 24 and 48 h. This can be explained by the reduced diffusive uptake of PEGNIO/DOX in comparison to free DOX (Fig. 3a and b).49,51 Due to the conjugation of the targeting ligand to PEGNIO/DOX, PEGNIO/DOX/CysTAT–MUC1 was more toxic to HeLa cells than to U87 cells after 24 and for 48 h. Both, after 24 and 48 hours PEGNIO/DOX/CysTAT–MUC1 had less cytotoxic effect of on U87 cells in comparison with free DOX (p < 0.01 and p < 0.05 respectively). PEGNIO/DOX/CysTAT–MUC1 increased the cytotoxicty for HeLa cells in comparison to PEGNIO/DOX for 24 and 48 hours (p < 0.01 and p < 0.05 respectively). Moreover in comparison to free DOX, PEGNIO/DOX/CysTAT–MUC1 showed a significantly increased toxic effect on HeLa cells after 48 h. According to obtained results, it is clear that the aptamer conjugated niosomal formulation acted as a targeted DOX delivery platform for MUC1 expressing tumor cells. | ||
| Fig. 5 Cytotoxicity of the free drug and niosomal formulations on HeLa and U87 cells. Cells were incubated with PEG/NIO, PEGNIO/DOX, PEGNIO/DOX/CysTAT–MUC1 and free DOX (equivalent concentration of loaded DOX) for 24 h (a) and for 48 h (b). MTT assay was applied. Error bars represent the standard deviation from the mean (N = 3). Data were analyzed using paired t-test, and *p < 0.05, **p < 0.01 was considered significant and very significant respectively. | ||
Conclusions
The objective of this study was to develop an efficient aptamer targeted niosomal drug delivery system. For this aim, PEGNIO was successfully synthesized by the thin film hydration method. The model drug DOX was encapsulated into the vesicles, and the surface of the vesicles was decorated with cell penetrating peptides and MUC1 aptamer as a targeting ligand. The drug-loaded niosomes exhibit great potential as targeting drug carriers. The targeted drug-loaded nanoparticles show stronger cytotoxicity of the MUC1 receptor overexpressed HeLa cells. As a conclusion, the formulation PEGNIO/DOX/CysTAT–MUC1 might be a promising and efficient strategy for the delivery of DOX to MUC1 overexpressed tumor cells.Acknowledgements
Konrad Adenauer Foundation is acknowledged for the financial support to Didem Ag Seleci. Martina Weiß and Rebecca Joncyzk (Leibniz University of Hannover, Institute for Technical Chemistry) are also acknowledged for technical help during HPLC and PCR analysis.Notes and references
- L. Brannon-Peppas and J. O. Blanchette, Adv. Drug Delivery Rev., 2004, 64, 1649–1659 CrossRef PubMed.
- A. Sankhyan and P. Pawar, J. Appl. Pharm. Sci., 2012, 02, 20–32 Search PubMed.
- D. Ag Seleci, M. Seleci, J.-G. Walter, F. Stahl and T. Scheper, J. Nanomater., 2016, 13 Search PubMed.
- F. B. Barlas, B. Demir, E. Guler, A. M. Senisik, H. A. Arican, P. Unak and S. Timur, RSC Adv., 2016, 6, 30217–30225 RSC.
- M. Hong, S. Zhu, Y. Jiang, G. Tang and Y. Pei, J. Controlled Release, 2009, 133, 96–102 CrossRef CAS PubMed.
- R. Muzzalupo, L. Tavano and C. La Mesa, Int. J. Pharm., 2013, 458, 224–229 CrossRef CAS PubMed.
- L. Tavano, R. Muzzalupo, L. Mauro, M. Pellegrino, S. Ando and N. Picci, Langmuir, 2013, 29, 12638–12646 CrossRef CAS PubMed.
- J. Park, P. M. Fong, J. Lu, K. S. Russell, C. J. Booth, W. M. Saltzman and T. M. Fahmy, Nanomedicine, 2009, 5, 410–418 CAS.
- A. I. Minchinton and I. F. Tannock, Nat. Rev. Cancer, 2006, 6, 583–592 CrossRef CAS PubMed.
- E. Koren and V. P. Torchilin, Trends Mol. Med., 2012, 18, 385–393 CrossRef CAS PubMed.
- W. L. L. Munyendo, H. Lv, H. Benza-Ingoula, L. D. Baraza and J. Zhou, Biomolecules, 2012, 2, 187–202 CrossRef CAS PubMed.
- M. Lindgren, K. Rosenthal-Aizman, K. Saar, E. Eiriksdottir, Y. Jiang, M. Sassian, P. Östlund, M. Hällbrink and Ü. Langel, Biochem. Pharmacol., 2006, 71, 416–425 CrossRef CAS PubMed.
- M. Seleci, D. A. Seleci, M. Ciftci, D. Odaci Demirkol, F. Stahl, S. Timur, T. Scheper and Y. Yagci, Langmuir, 2015, 31, 4542–4551 CrossRef CAS PubMed.
- S. Deshayes, M. C. Morris, G. Divita and F. Heitz, Cell. Mol. Life Sci., 2005, 62, 1839–1849 CrossRef CAS PubMed.
- M. Mano, C. Teodosio, S. Simoes, d. L. M. Pedroso and A. Paiva, Biochem. J., 2005, 390, 603–612 CrossRef CAS PubMed.
- N. Schmidt, A. Mishra, G. H. Lai and G. C. L. Wong, FEBS Lett., 2010, 584, 1806–1813 CrossRef CAS PubMed.
- B. R. Liu, Y.-W. Huang, H.-J. Chiang and H.-J. Lee, J. Nanosci. Nanotechnol., 2010, 10, 7897–7905 CrossRef CAS PubMed.
- E. Vives, J. Schmidt and A. Pelegrin, Biochim. Biophys. Acta, Rev. Cancer, 2008, 1786, 126–138 CrossRef CAS PubMed.
- J. Taylor-Papadimitriou, J. Burchell, D. W. Miles and M. Dalziel, Biochim. Biophys. Acta, Mol. Basis Dis., 1999, 1455, 301–313 CrossRef CAS.
- Z. Cao, R. Tong, A. Mishra, W. Xu, G. C. L. Wong, J. Cheng and Y. Lu, Angew. Chem., Int. Ed., 2009, 48, 6494–6498 CrossRef CAS PubMed.
- T. Chen, M. I. Shukoor, Y. Chen, Q. Yuan, Z. Zhu, Z. Zhao, B. Gulbakan and W. Tan, Nanoscale, 2011, 3, 546–556 RSC.
- Z. Liu, H. Zhao, L. He, Y. Yao, Y. Zhou, J. Wu, J. Liu and J. Ding, RSC Adv., 2015, 5, 16931–16939 RSC.
- A. Ozer, J. M. Pagano and J. T. Lis, Mol. Ther.--Nucleic Acids, 2014, 3, e183 CrossRef CAS PubMed.
- C. S. M. Ferreira, C. S. Matthews and S. Missailidis, Tumor Biol., 2006, 27, 289–301 CrossRef CAS PubMed.
- Y. Hu, J. Duan, Q. Zhan, F. Wang, X. Lu and X.-D. Yang, PLoS One, 2012, 7, e31970 CAS.
- C. Yu, Y. Hu, J. Duan, W. Yuan, C. Wang, H. Xu and X.-D. Yang, PLoS One, 2011, 6, e24077 CAS.
- L. Tan, K. G. Neoh, E. T. Kang, W. S. Choe and X. Su, Macromol. Biosci., 2011, 11, 1331–1335 CrossRef CAS PubMed.
- M. N. Azmin, A. T. Florence, R. M. Handjani-Vila, J. F. B. Stuart, G. Vanlerberghe and J. S. Whittaker, J. Pharm. Pharmacol., 1985, 37, 237–242 CrossRef CAS PubMed.
- Z.-X. Liao, E.-Y. Chuang, C.-C. Lin, Y.-C. Ho, K.-J. Lin, P.-Y. Cheng, K.-J. Chen, H.-J. Wei and H.-W. Sung, J. Controlled Release, 2015, 208, 42–51 CrossRef CAS PubMed.
- J. Takasaki and S. M. Ansell, Bioconjugate Chem., 2006, 17, 438–450 CrossRef CAS PubMed.
- L. Tavano, R. Aiello, G. Ioele, N. Picci and R. Muzzalupo, Colloids Surf., B, 2014, 118, 7–13 CrossRef CAS PubMed.
- S. B. Shirsand, M. S. Para, D. Nagendrakumar, K. M. Kanani and D. Keerthy, Int. J. Pharm. Invest., 2012, 2, 201–207 CrossRef CAS PubMed.
- G. Wei, S. Xiao, D. Si and C. Liu, Biomed. Chromatogr., 2008, 22, 1252–1258 CrossRef CAS PubMed.
- A. Shrivastava and V. B. Gupta, Chron. Young Sci., 2011, 2, 21–25 CrossRef.
- R. Bongartz, D. Ag, M. Seleci, J.-G. Walter, E. E. Yalcinkaya, D. O. Demirkol, F. Stahl, S. Timur and T. Scheper, J. Mater. Chem. B, 2013, 1, 522–528 RSC.
- G. Minotti, P. Menna, E. Salvatorelli, G. Cairo and L. Gianni, Pharmacol. Rev., 2004, 56, 185–229 CrossRef CAS PubMed.
- P. Mohan and N. Rapoport, Mol. Pharmaceutics, 2010, 7, 1959–1973 CrossRef CAS PubMed.
- B. Haley and E. Frenkel, Urologic Oncology: Seminars and Original Investigations, 2008 Search PubMed.
- T. M. Allen and P. R. Cullis, Science, 2004, 303, 1818–1822 CrossRef CAS PubMed.
- G. T. Hermanson, Bioconjugate Techniques, Academic press, 2nd edn, 2008 Search PubMed.
- Y. Li, M. Kröger and W. K. Liu, Biomaterials, 2014, 35, 8467–8478 CrossRef CAS PubMed.
- J. V. Jokerst, T. Lobovkina, R. N. Zare and S. S. Gambhir, Nanomedicine, 2011, 6, 715–728 CrossRef CAS PubMed.
- N. O. Sahin, in Nanomaterials and Nanosystems for Biomedical Applications, Springer, 2007, pp. 67–81 Search PubMed.
- M. Bragagni, N. Mennini, C. Ghelardini and P. Mura, J. Pharm. Pharm. Sci., 2012, 15, 184–196 CAS.
- G. A. Hughes, Nanomedicine, 2005, 1, 22–30 CAS.
- J. Qi, P. Yao, F. He, C. Yu and C. Huang, Int. J. Pharm., 2010, 393, 177–185 CrossRef PubMed.
- C. t. S. M. Ferreira, M. C. Cheung, S. Missailidis, S. Bisland and J. Gariepy, Nucleic Acids Res., 2009, 37, 866–876 CrossRef CAS PubMed.
- M. S. Syrkina, M. A. Rubtsov, D. M. Potashnikova, Y. D. Kondratenko, A. A. Dokrunova and V. P. Veiko, Acta Naturae, 2014, 6, 62–70 CAS.
- X.-B. Xiong, Y. Huang, W.-l. Lu, X. Zhang, H. Zhang, T. Nagai and Q. Zhang, J. Controlled Release, 2005, 107, 262–275 CrossRef CAS PubMed.
- A. D. Heibein, B. Guo, J. A. Sprowl, D. A. MacLean and A. M. Parissenti, BMC Cancer, 2012, 12, 381 CrossRef CAS PubMed.
- X. Li, L. Ding, Y. Xu, Y. Wang and Q. Ping, Int. J. Pharm., 2009, 373, 116–123 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra19525c |
| This journal is © The Royal Society of Chemistry 2016 |