
Discovery of cytochrome bc1 complex inhibitors inspired by the natural product karrikinolide†
Cheng Chenb,
Qiong-You Wu*a,
Lian-Ying Shana,
Bei Zhanga,
Francis Verpoortb and
Guang-Fu Yang*a
aKey Laboratory of Pesticide & Chemical Biology, College of Chemistry, Central China Normal University, Wuhan 430079, P. R. China. E-mail: qywu@mail.ccnu.edu.cn; gfyang@mail.ccnu.edu.cn; Fax: +86-27-67867141; Tel: +86-27-67867706
bState Key Laboratory of Advanced Technology for Materials Synthesis and Processing, Wuhan University of Technology, Wuhan 430070, P. R. China
First published on 5th October 2016
Abstract
The cytochrome bc1 complex (cyt bc1 or complex III) is a promising target of numerous antibiotics and fungicides. With an aim to indentify new lead structures for the bc1 complex, a series of novel inhibitors were discovered from the natural product karrikinolide for the first time. Extensive screening results suggested variable inhibitory activities of these compounds against succinate-cytochrome reductase [SCR, a mixture of respiratory complex II (SQR) and complex III (the bc1 complex)], implying the essential role of a 4-substituted phenyl group for the high potency. Exceptionally, compound 12g showed excellent inhibition potency having an IC50 value in the sub-micromolar range, demonstrating its higher potency than the commercial control amisulbrom by over two orders of magnitude. Further experiments inferred that these newly prepared compounds mainly target the bc1 complex. Seemingly, this work has presented a new lead scaffold for further development of bc1 complex inhibitors.
1. Introduction
Oxidative phosphorylation and the tricarboxylic acid (or Krebs) cycle, two bioprocesses that are coupled with electron transport and proton pump flow, constitute the highly effective mitochondrial respiration. In animals and bacteria, the oxidative phosphorylation system comprises five multiprotein complexes (complexes I, II, III, IV, and V) and two mobile electron carriers (ubiquinone and cytochrome c) embedded in the lipid bilayer of the mitochondrial inner membrane.1 Among them, cytochrome bc1 complex (EC 1.10.2.2, bc1) is an essential component of the cellular respiratory chain and plays a pivotal role in the life cycle. The bc1 complex catalyzes the electron transfer from ubiquinol (hydroxyquinone, QH2) to a water-soluble cytochrome c (cyt c) and couples this electron transfer to the translocation of protons across the membrane, generating a proton gradient and membrane potential for ATP synthesis.2–5 In case the electron transport process of the bc1 complex is disturbed or disrupted, the cellular respiration will be blocked, resulting in cell death. Therefore, the bc1 complex is one of the most attractive targets for the development of pharmaceuticals and agrochemicals. Over the past years, pronounced attention has been shown for the development of cytochrome bc1 complex inhibitors,6–17 and our group has also been engaged extensively in this area.1,18–23 Although numerous inhibitors have been identified, the discovery of novel inhibitors with entirely new scaffolds still attracts growing attention.Normally, natural products show a broad range of interesting and useful biological activities. More significantly, they demonstrate excellent environmental compatibility and could be readily biodegraded.24–26 However, limited activities and adverse photo and chemical stabilities of these compounds often indulge the use of their synthetic analogues instead,27 which usually serve as ideal candidates for the development of medicines and pesticides.28,29 Through structural modifications, these analogues could give rise to novel structures with improved biological activities, new modes of action, and increased safety towards living beings and the environment. Consequently, structural modifications inspired from natural products are an ideal approach to identify new lead molecules with useful functionalities.30
To date, notable examples of the successful utilization of natural products as commercial inhibitors of the cytochrome bc1 complex are analogues of the naturally occurring β-methoxyacrylate strobilurin A.31–33 In addition, structural optimizations based on antimycin A,34–37 neopeltolide,38–44 crocacin D45–48 have also been extensively reported. Despite enormous efforts devoted to the discovery of natural product-derived inhibitors, there is still a high demand for identification of new lead structures from natural products which contain totally different structures from existing inhibitors. Karrikinolide, a natural product bearing a 2H-furo[2,3-c]pyran-2-one motif, captured our attention due to its molecular simplicity, structural stability, novel skeleton, as well as diverse bioactivities including its incredible potency for promoting seed germination and seedling development of crop plants,49–62 and antibacterial activities.63–66 It has been demonstrated that substituents on the C-3 position played a significant role on the biological activities of the corresponding compounds.55,67–69
Therefore, our goal was to synthesize a multitude of karrikinolide derivatives by introducing diverse functional groups on the C-3 position with the aim to adjust the global molecular physicochemical characteristics (Scheme 1). Afterwards, as a continuation of our research on the development of novel cytochrome bc1 complex inhibitors, the novel karrikinolide derivatives were extensively screened on their inhibiting performance towards the cytochrome bc1 complex. Hence, it was discovered, for the first time, that some of these compounds exhibit high inhibitory activities.
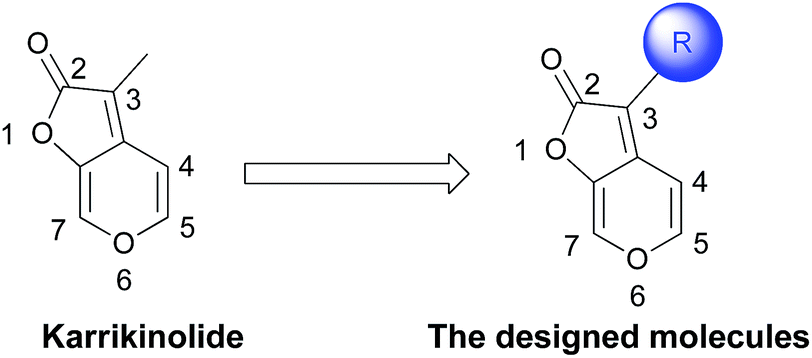 | ||
| Scheme 1 Karrikinolide and the designed molecules. | ||
2. Results and discussion
It has been well-documented that karrikinolide was originally isolated from plant smoke.49,50 However, it is insufficient to obtain this compound merely in this way. Consequently, several research groups developed their respective total synthesis of karrikinolide e.g. via the intramolecular Wittig reaction with thioketone, Ti-crossed aldol addition and Cu(II)-catalyzed lactonization.70–74 Among these approaches, Goddard-Borger's protocol achieved the target karrikinolide in relatively higher yield.71 In particular, this approach takes advantage of readily available D-xylose (1) to construct the 2H-furo[2,3-c]pyran-2-one 2 which, we realized, would allow us to investigate the effect of modifications at this position on the activity of karrikinolide analogues (Scheme 2). As depicted in Scheme 2, the key intermediate 2 in our envisioned approach could be obtained from D-xylose (1) after a few steps. Afterwards, 2 could be converted to the corresponding aldehyde 3, which could be further transformed to the natural product karrikinolide. Alternatively, 2 could react with NBS to give the bromide 4, which would undergo palladium-catalyzed cross coupling reactions for formation of our designed target molecules. | ||
| Scheme 2 Synthetic route to karrikinolide and the designed molecules. | ||
The detailed synthetic procedure for the synthesis of karrikinolide and its analogues could be described in Scheme 3. The first step is the straightforward generation of compound 5 from 1, followed by the regioselective tritylation of 5 to give the alcohol 6. Oxidation of 6 afforded the ketone 7, which was transformed to the desired (Z)-olefin 8 in high yield by utilizing the Horner–Wadsworth–Emmons methodology. Treatment of 8 with aqueous trifluoroacetic acid resulted in rapid formation of the butenolide 9 in excellent yield via hydrolysis, lactonisation and rearrangement. Afterwards, 9 was treated with acetic anhydride in pyridine to exclusively yield the α-diacetate 10, which went through an elimination to give 11 in the presence of triethylamine. Indeed, treatment of 11 with Pd(PPh3)4 in refluxing THF provided the key intermediate 2. Thereafter two reaction directions are possible using 2 – it would either form the aldehyde 3, which could subsequently undergo a reductive deoxygenation to give the natural product karrikinolide, or would be brominated to the corresponding bromide 4, which could be further functionalized to a variety of karrikinolide derivatives.
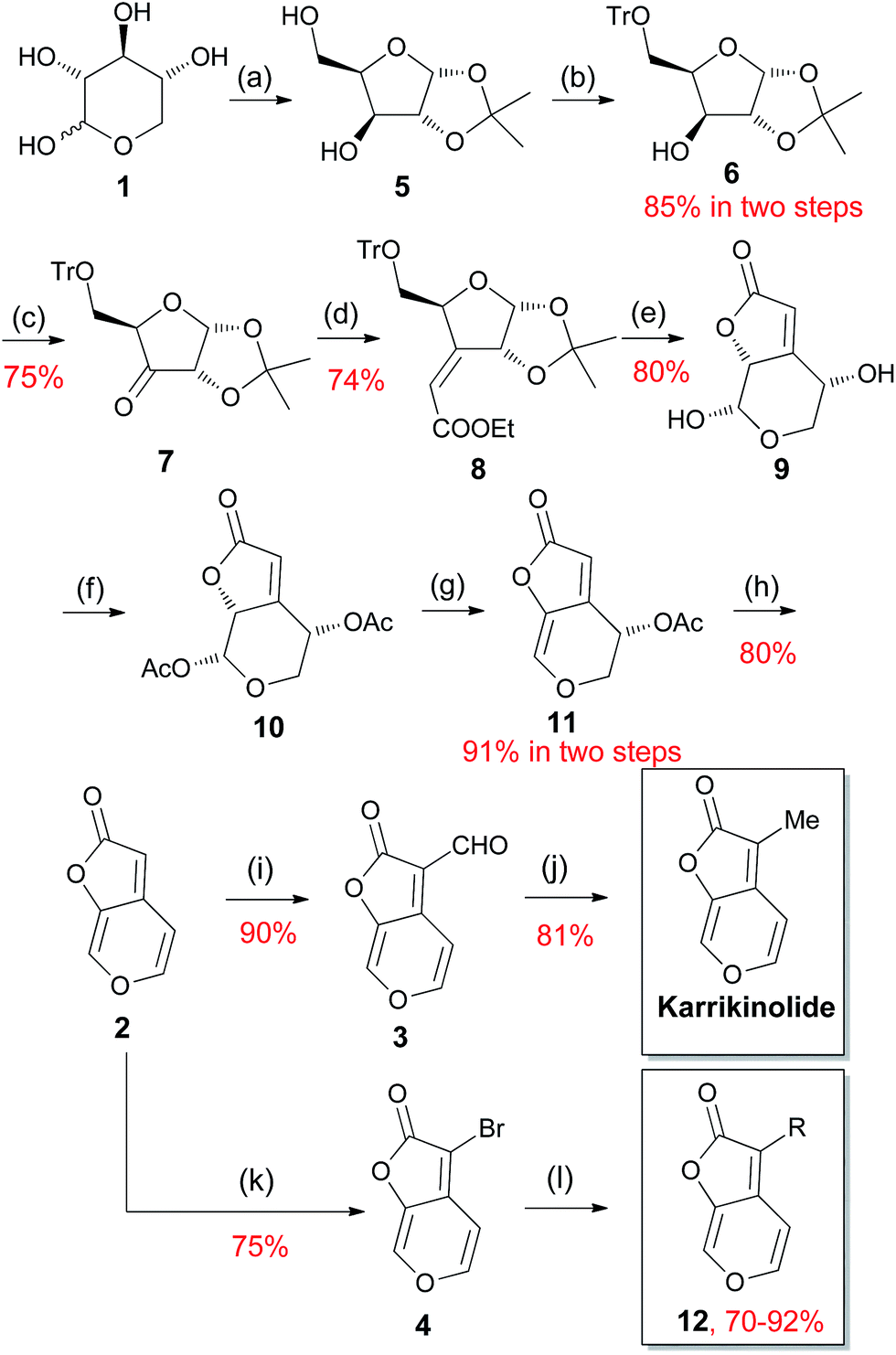 | ||
| Scheme 3 The designed synthetic route of karrikinolide and its analogues. Reagents and conditions: (a) (1) CuSO4, H2SO4, acetone, rt, 12 h, (2) aq. HCl, rt, 15 h; (b) (1) Et3N, CH2Cl2, rt, 1 h; (2) CPh3Cl, rt, 3 h; (c) Ac2O, DMSO, rt, 22 h; (d) NaH, (EtO)2POCH2CO2Et, THF, −10 °C, 1.5 h; (e) CF3COOH, H2O, rt, 30 min; (f) Ac2O, C5H5N, rt, 2 h; (g) Et3N, CH2Cl2, 30 min; (h) Pd(PPh3)4, THF, reflux, 48 h; (i) (1) POCl3, DMF, 50 °C, (2) sat. aq. NaHCO3; (j) tBuNH2·BH3, AlCl3, CH2Cl2, reflux; (k) NBS, MeOH, rt, 20 min; (l) R-Bu(OH)2, Pd(OAc)2, SPhos, K3PO4, toluene, reflux. | ||
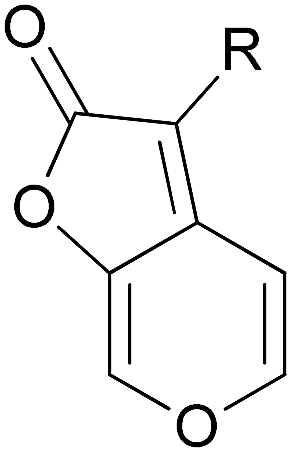
Succinate-cytochrome reductase (SCR) is composed of respiratory complex II (SQR) and complex III (the bc1 complex), and are believed to form a complex II–complex III supercomplex.75 Inhibitory activities of the newly prepared compounds against SCR were tested at a concentration of 100 μM and listed in Table 1. The IC50 values of the compounds with inhibition activities higher than 50% were further determined. The first and foremost, the inhibitory activity of karrikinolide was tested against SCR. Unfortunately, an inhibition ratio of 13% was obtained at the concentration of 100 μM (entry 1). Meanwhile, low inhibition rates of compounds 2–4 demonstrated that the less bulky groups such as H, –CHO and Br were unfavorable (entries 2–4). To our delight, when a phenyl group was employed as R, an inhibition rate of 52% was observed (entry 5). This result inspired us to conduct further structural optimizations via implementation of different substituents on the phenyl ring. Therefore, substituted phenyl groups were then introduced for R. The results implied that employment of a substituent at the para-position of the phenyl ring resulted in significant improvement of the inhibitory activities in most cases (entries 6–13). Apparently, alkyl substituents bearing hydrophobic linear chains are superior to other functional groups such as halogen and ethoxyl. Notably, a sub-micromolar inhibitor, compound 12g with 4-n-butylphenyl as R, was identified as being over 100-fold more active compared to the commercial control amisulbrom (IC50 = 0.737 μM for entry 11 vs. IC50 = 93.0 μM for entry 22). Replacing the n-butyl group at the para-position on the phenyl ring with sterically hindered alkyl substituents reduced the potency of the resulting compounds, as observed for the i-butyl 12h and t-butyl 12i derivatives. The i-butyl analogue 12h (IC50 = 1.1 μM) demonstrated comparable inhibitory activity in comparison to the counterpart n-butyl inhibitor, while the t-butyl analogue (IC50 = 10.7 μM) was at least 10 times less potent than the n-butyl-bearing derivative. In addition, a substituent at either the meta- or ortho-position of the phenyl ring didn't significantly affect the activities (entries 14 and 15), with respect to the 49% and 46% inhibition rates of compounds 12j and 12k in which a 3-methoxyl or 2-chloro substituent was introduced, respectively. Furthermore, poly-substituted phenyls were also evaluated. It appears that poly-substitution on the phenyl ring is detrimental to the inhibition potency. Compounds 12l–12o manifested inhibition rates ranging from 25–47% at the concentration of 100 μM (entries 16–19). Compounds of which the phenyl ring is replaced with a heterocyclic ring (either furyl or pyridyl), compounds 12p and 12q respectively, produced relatively poor levels of inhibition activities, demonstrating inhibition potency of less than 10% (entries 20 and 21).
|
|||
|---|---|---|---|
| Entry | Compound no. | R | Inhibition ratio (100 μM)/IC50 (μM) |
| a The inhibition ratio was determined at the concentration of 100 μM.b IC50 values. | |||
| 1 | Karrikinolide | –Me | 13%a |
| 2 | 2 | –H | <10%a |
| 3 | 3 | –CHO | <10%a |
| 4 | 4 | –Br | 21%a |
| 5 | 12a | C6H5– | 52%a |
| 6 | 12b | 4-Cl-C6H4– | 26.90 ± 0.16b |
| 7 | 12c | 4-CF3O-C6H4– | 3.55 ± 0.11b |
| 8 | 12d | 4-EtO-C6H4– | 50%a |
| 9 | 12e | 4-iPrO-C6H4– | 9.39 ± 0.12b |
| 10 | 12f | 4-nPr-C6H4– | 8.50 ± 0.13b |
| 11 | 12g | 4-nBu-C6H4– | 0.737 ± 0.011b |
| 12 | 12h | 4-iBu-C6H4– | 1.10 ± 0.20b |
| 13 | 12i | 4-tBu-C6H4– | 10.71 ± 0.10b |
| 14 | 12j | 3-MeO-C6H4– | 49%a |
| 15 | 12k | 2-Cl-C6H4– | 46%a |
| 16 | 12l | 2-C12H7– | 47%a |
| 17 | 12m | 2,4-Di-Cl-C6H3– | 41%a |
| 18 | 12n | 4-Me-2-Cl-C6H3– | 30%a |
| 19 | 12o | 3,5-Di-F-C6H3– | 25%a |
| 20 | 12p |  |
<10%a |
| 21 | 12q |  |
<10%a |
| 22 | Amisulbrom | 93.0 ± 1.3b | |
Since SCR is composed of respiratory complex II (SQR) and complex III (cyt bc1), it is of significant interest and importance to further determine whether the synthesized compounds are inhibitors targeting SQR or the bc1 complex. As listed in Table 2, we compared the activities of four representative compounds (12c, 12e, 12g and 12h) against SCR, SQR and cyt bc1. The results indicated that all these compounds exhibited limited activity against SQR at a concentration of 10 μM. Moreover, their inhibitory activities against SCR and the bc1 complex were comparable at the same concentration. Therefore, it can be concluded from the above results that the target compounds in this article have been confirmed as inhibitors of the bc1 complex.
| Compound no. | IC50 (μM), SCR | I% (10 μM), SCR | I% (10 μM), SQR | I% (10 μM), cyt bc1 | Selectivity |
|---|---|---|---|---|---|
| 12c | 3.55 ± 0.11 | 76% | 14% | 66% | cyt bc1 |
| 12e | 9.39 ± 0.12 | 50% | <10% | 46% | cyt bc1 |
| 12g | 0.737 ± 0.011 | 82% | 39% | 83% | cyt bc1 |
| 12h | 1.10 ± 0.20 | 73% | 26% | 67% | cyt bc1 |
3. Conclusion
A new series of 2H-furo[2,3-c]pyran-2-one derivatives were designed and synthesized via structural modifications from the natural product karrikinolide. It is worth mentioning that this is the first time to discover bc1 complex inhibitors based on karrikinolide. The bioassay results indicated that these newly prepared compounds exhibited varied inhibition against SCR, and a 4-substituted phenyl group as the R substituent is paramount for maintaining high potency. Excitingly, compound 12g showed inhibition potency at the sub-micromolar level, which demonstrated over 100-fold higher activity than amisulbrom, a commercial inhibitor of the bc1 complex. Further evaluation against the respective SQR and the bc1 complex indicated that these compounds are inhibitors of the bc1 complex. Consequently, a totally new skeleton that is different from existing bc1 complex inhibitors was discovered, which could provide a new lead for further development of bc1 complex inhibitors.4. Experimental section
4.1 General considerations
1H-NMR spectra were recorded on a VARIAN Mercury-Plus 600 or 400 spectrometer or a Bruker Avance 500 spectrometer in CDCl3 or (CD3)2CO with TMS as the internal reference. 13C-NMR spectra were recorded in CDCl3 or (CD3)2CO on a VARIAN Mercury-Plus 600 (150 MHz) or 400 (100 MHz) spectrometer or a Bruker Avance 500 (125 MHz) spectrometer. The following abbreviations are used to designate multiplicities: s = singlet, d = doublet, dd = doublet of doublets, t = triplet, q = quartet, m = multiplet. HR-MS was taken on an Agilent 6520 Accurate-Mass Q-TOF instrument or a Bruker Daltonics microTOF-QII instrument. Melting points were taken on a Buchi B-545 melting point apparatus and are uncorrected. Reagents including D-xylose (1) and solvents were commercially available and treated with standard methods before use. Besides, 2H-furo[2,3-c]pyran-2-one (2),71 2-oxo-2H-furo[2,3-c]pyran-3-carbaldehyde (3),71 3-bromo-2H-furo[2,3-c]pyran-2-one (4),72 5-(hydroxymethyl)-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-6-ol (5),76 2,2-dimethyl-5-((trityloxy)methyl)tetrahydrofuro[2,3-d][1,3]dioxol-6-ol (6),77 2,2-dimethyl-5-((trityloxy)methyl)dihydrofuro[2,3-d][1,3]dioxol-6(3aH)-one (7),77 (Z)-ethyl 2-(2,2-dimethyl-5-((trityloxy)methyl)furo[2,3-d][1,3]dioxol-6(3aH,5H,6aH)-ylidene) acetate (8),71 4,7-dihydroxy-4,5,7,7a-tetrahydro-2H-furo[2,3-c]pyran-2-one (9),71 2-oxo-4,5,7,7a-tetrahydro-2H-furo[2,3-c]pyran-4,7-diyl diacetate (10),71 2-oxo-4,5-dihydro-2H-furo[2,3-c]pyran-4-yl acetate (11),71 and karrikinolide71 were prepared according to literature reports.4.2 General procedure for synthesis of compounds 12a–12q
Inside an argon-filled glove box, Pd(OAc)2 (0.03 mmol), 2-dicyclohexylphosphino-2′,6′-dimethoxybiphenyl (SPhos, 0.075 mmol), compound 4 (1.0 mmol), K3PO4 (2.0 mmol), a boronic acid (1.50 mmol) and toluene (5 mL) were added to an oven-dried Schlenk tube. The Schlenk tube was taken out of the glove box and the mixture was heated to reflux under argon atmosphere for 24 h. The reaction mixture was cooled down to room temperature, and the solvent was removed in vacuo. The residue was purified by silica gel flash column chromatography to afford the desired products 12a–12q.4.3 Analytical data for karrikinolide, compounds 2–4 and 12a–12q
4.4 Enzyme assay
The overall activity of SCR (both SQR and cyt bc1) was determined using succinate and cytochrome c as substrates. The activity of SQR in SCR was selectively determined by using succinate and dichlorophenolindophenol (DCIP) as substrates, while that of cyt bc1 in SCR was determined by using decylubiquinol (DBH2) and cytochrome c as substrates.75,78 The preparation of SCR from the porcine heart was essential as reported,1 and the preparation of DBH2 from DB was carried out according to the procedure described in a previous publication.75 To obtain the IC50 values of the compounds, all reactions were initiated by the addition of enzyme and monitored continuously by following the absorbance change at certain wavelengths on a PerkinElmer Lambda 45 spectrophotometer equipped with a magnetic stirrer at 23 °C.78The activity of SCR was measured by monitoring the increase of cytochrome c at 550 nm, by using the extinction coefficient of 18.5 mM−1 cm−1. The activity of SQR was measured by monitoring the decrease of 2,6-dichlorophenolindophenol (DCIP) at 600 nm, by using the extinction coefficient of 21 mM−1 cm−1. The reaction mixture may be scaled down to 1.8 mL with final concentrations of PBS (pH 7.4), 100 mM; EDTA, 0.3 mM; succinate, 20 mM; oxidized cytochrome c, 60 mM (or DCIP, 53 mM); and appropriate amounts of enzyme to start the reaction. Moreover, the activity of cyt bc1 in catalyzing the oxidation of DBH2 by cytochrome c was measured by monitoring the increase of cytochrome c at 550 nm, and assayed in 100 mM PBS (pH 7.4), 0.3 mM EDTA, 750 μM lauryl maltoside (n-dodecyl-b-D-maltoside), 100 μM DBH2, 100 μM oxidized cytochrome c, and an appropriate amount of SCR. The detailed description for performing the enzyme assay could be referred to our previous publications.1,18–23
Acknowledgements
This research was supported by the National Key Technologies R&D Program (2014BAD23B01) and the National Natural Science Foundation of China (No. 21272091, 21332004, 21472063).Notes and references
- H. Cheng, Y.-Q. Shen, X.-Y. Pan, Y.-P. Hou, Q.-Y. Wu and G.-F. Yang, New J. Chem., 2015, 39, 7281–7292 RSC.
- D. Xia, L. Esser, L. Yu and C.-A. Yu, Photosynth. Res., 2007, 92, 17–34 CrossRef CAS PubMed.
- A. R. Crofts, Annu. Rev. Physiol., 2004, 66, 689–733 CrossRef CAS PubMed.
- H. Kim, D. Xia, C. A. Yu, J. Z. Xia, A. M. Kachurin, L. Zhang, L. Yu and J. Deisenhofer, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 8026–8033 CrossRef CAS.
- E. A. Berry, M. Guergova-Kuras, L. S. Huang and A. R. Crofts, Annu. Rev. Biochem., 2000, 69, 1005–1075 CrossRef CAS PubMed.
- E. C. Slater, Biochim. Biophys. Acta, Rev. Bioenerg., 1973, 301, 129 CrossRef CAS.
- J. M. Clough, C. R. A. Godfrey, J. R. Godwin, R. S. I. Joseph and C. Spinks, Pestic. Outlook, 1996, 7, 16 CAS.
- H. L. Ypema and R. E. Gold, Plant Dis., 1999, 83, 4 CrossRef CAS.
- M. Ueki, K. Machida and M. Taniguchi, Curr. Opin. Anti-Infect. Invest. Drugs, 2000, 2, 387 CAS.
- S. Mitani, S. Araki, Y. Takii, T. Ohshima, N. Matsuo and H. Miyoshi, Pestic. Biochem. Physiol., 2001, 71, 107–115 CrossRef CAS.
- T. Ohshima, T. Komyoji, S. Mitani, N. Matsuo and T. Nakajima, J. Pestic. Sci., 2004, 29, 136–138 CrossRef.
- S. Bolgunas, D. A. Clark, W. S. Hanna, P. A. Mauvais and S. O. Pember, J. Med. Chem., 2006, 49, 4762–4766 CrossRef CAS PubMed.
- A. G. Kruglov, M. A. Andersson, R. Mikkola, M. Roivainen, L. Kredics, N.-E. L. Saris and M. S. Salkinoja-Salonen, Chem. Res. Toxicol., 2009, 22, 565–573 CrossRef CAS PubMed.
- E. A. Berry, L.-s. Huang, D.-W. Lee, F. Daldal, K. Nagai and N. Minagawa, Biochim. Biophys. Acta, Bioenerg., 2010, 1797, 360–370 CrossRef CAS PubMed.
- C. Vallieres, N. Fisher, T. Antoine, M. Al-Helal, P. Stocks, N. G. Berry, A. S. Lawrenson, S. A. Ward, P. M. O'Neill, G. A. Biagini and B. Meunier, Antimicrob. Agents Chemother., 2012, 56, 3739–3747 CrossRef CAS PubMed.
- M. J. Capper, P. M. O'Neill, N. Fisher, R. W. Strange, D. Moss, S. A. Ward, N. G. Berry, A. S. Lawrenson, S. S. Hasnain, G. A. Biagini and S. V. Antonyuk, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 755–760 CrossRef CAS PubMed.
- M. Fehr, A. Wolf and G. Stammler, Pest Manage. Sci., 2016, 72, 591–602 CrossRef CAS PubMed.
- P.-L. Zhao, L. Wang, X.-L. Zhu, X. Huang, C.-G. Zhan, J.-W. Wu and G.-F. Yang, J. Am. Chem. Soc., 2010, 132, 185–194 CrossRef CAS PubMed.
- F. Wang, H. Li, L. Wang, W.-C. Yang, J.-W. Wu and G.-F. Yang, Bioorg. Med. Chem., 2011, 19, 4608–4615 CrossRef CAS PubMed.
- G.-F. Hao, F. Wang, H. Li, X.-L. Zhu, W.-C. Yang, L.-S. Huang, J.-W. Wu, E. A. Berry and G.-F. Yang, J. Am. Chem. Soc., 2012, 134, 11168–11176 CrossRef CAS PubMed.
- W. C. Yang, H. Li, F. Wang, X. L. Zhu and G. F. Yang, ChemBioChem, 2012, 13, 1542–1551 CrossRef CAS PubMed.
- X. Zhu, F. Wang, H. Li, W. Yang, Q. Chen and G. Yang, Chin. J. Chem., 2012, 30, 1999–2008 CrossRef CAS.
- X. Zhu, M. Zhang, J. Liu, J. Ge and G. Yang, J. Agric. Food Chem., 2015, 63, 3377–3386 CrossRef CAS PubMed.
- D. J. Newman, G. M. Cragg and K. M. Snader, J. Nat. Prod., 2003, 66, 1022–1037 CrossRef CAS PubMed.
- D. J. Newman and G. M. Cragg, J. Nat. Prod., 2012, 75, 311–335 CrossRef CAS PubMed.
- D. J. Newman and G. M. Cragg, J. Nat. Prod., 2016, 79, 629–661 CrossRef CAS PubMed.
- A. Kumar, S. Srivastava, G. Gupta, V. Chaturvedi, S. Sinha and R. Srivastava, ACS Comb. Sci., 2011, 13, 65–71 CrossRef CAS PubMed.
- L. O. Haustedt, C. Mang, K. Siems and H. Schiewe, Curr. Opin. Drug Discovery Dev., 2006, 9, 445–462 CAS.
- D. J. Newman and G. M. Cragg, J. Nat. Prod., 2007, 70, 461–477 CrossRef CAS PubMed.
- C. Cordier, D. Morton, S. Murrison, A. Nelson and C. O'Leary-Steele, Nat. Prod. Rep., 2008, 25, 719–737 RSC.
- H. Sauter, W. Steglich and T. Anke, Angew. Chem., Int. Ed., 1999, 38, 1328–1349 CrossRef.
- D. W. Bartlett, J. M. Clough, J. R. Godwin, A. A. Hall, M. Hamer and B. Parr-Dobrzanski, Pest Manage. Sci., 2002, 58, 649–662 CrossRef CAS PubMed.
- L. Lin, N. Mulholland, Q. Y. Wu, D. Beattie, S. W. Huang, D. Irwin, J. Clough, Y. C. Gu and G. F. Yang, J. Agric. Food Chem., 2012, 60, 4480–4491 CrossRef CAS PubMed.
- E. C. Slater, Biochim. Biophys. Acta, 1974, 301, 129–154 CrossRef.
- G. V. Jagow and T. A. Link, Methods Enzymol., 1986, 126, 253–271 Search PubMed.
- N. Tokutake, H. Miyoshi, H. Nakazato and H. Iwamura, Biochim. Biophys. Acta, 1993, 1142, 262–268 CrossRef CAS.
- D. Canter, S. Boorjian, K. Ogan, N. Shore, T. Bivalacqua, B. Bochner, T. Downs, L. Gomella, R. Grubb and B. Inman, J. Med. Chem., 2006, 49, 4762–4766 CrossRef PubMed.
- A. E. Wright, J. C. Botelho, E. Guzmán, D. Harmody, P. Linley, P. J. McCarthy, T. P. Pitts, S. A. Pomponi and J. K. Reed, J. Nat. Prod., 2007, 70, 412–416 CrossRef CAS PubMed.
- W. Youngsaye, J. T. Lowe, F. Pohlki, P. Ralifo and J. S. Panek, Angew. Chem., Int. Ed., 2007, 46, 9211–9214 CrossRef CAS PubMed.
- D. W. Custar, T. P. Zabawa and K. A. Scheidt, J. Am. Chem. Soc., 2008, 130, 804–805 CrossRef CAS PubMed.
- O. A. Ulanovskaya, J. Janjic, M. Suzuki, S. S. Sabharwal, P. T. Schumacker, S. J. Kron and S. A. Kozmin, Nat. Chem. Biol., 2008, 4, 418–424 CrossRef CAS PubMed.
- V. V. Vintonyak, B. Kunze, F. Sasse and M. E. Maier, Chem.–Eur. J., 2008, 14, 11132–11140 CrossRef CAS PubMed.
- Y. Cui, W. Tu and P. E. Floreancig, Tetrahedron, 2010, 66, 4867–4873 CrossRef CAS PubMed.
- H. Fuwa, T. Noguchi, M. Kawakami and M. Sasaki, Bioorg. Med. Chem. Lett., 2014, 24, 2415–2419 CrossRef CAS PubMed.
- B. Kunze, R. Jansen, G. Höfle and H. Reichenbach, J. Antibiot., 1994, 47, 881–886 CrossRef CAS PubMed.
- P. J. Crowley, E. A. Berry, T. Cromartie, F. Daldal, C. R. A. Godfrey, D.-W. Lee, J. E. Phillips, A. Taylor and R. Viner, Bioorg. Med. Chem., 2008, 16, 10345–10355 CrossRef CAS PubMed.
- C. F. Nising, S. Hillebrand and L. Rodefeld, Chem. Commun., 2011, 47, 4062–4073 RSC.
- A. E. Pasqua, F. D. Ferrari, J. J. Crawford, W. G. Whittingham and R. Marquez, Bioorg. Med. Chem., 2015, 23, 1062–1068 CrossRef CAS PubMed.
- G. R. Flematti, E. L. Ghisalberti, K. W. Dixon and R. D. Trengove, Science, 2004, 305, 977 CrossRef CAS PubMed.
- J. V. Staden, A. K. Jäger, M. E. Light, B. V. Burger, N. A. C. Brown and T. H. Thomas, S. Afr. J. Bot., 2004, 70, 654–659 CrossRef.
- G. R. Flematti, E. L. Ghisalberti, K. W. Dixon, R. D. Trengove, B. W. Skelton and A. H. White, Aust. J. Chem., 2005, 58, 505–506 CrossRef CAS.
- M. E. Light, B. V. Burger and J. van Staden, J. Agric. Food Chem., 2005, 53, 5936–5942 CrossRef CAS PubMed.
- N. Jain and J. Van Staden, Plant Growth Regul., 2006, 50, 139–148 CrossRef CAS.
- N. Jain, M. G. Kulkarni and J. Staden, Plant Growth Regul., 2006, 49, 263–267 CrossRef CAS.
- D. C. Nelson, F. A. Riseborough, G. R. Flematti, J. Stevens, E. L. Ghisalberti, K. W. Dixon and S. M. Smith, Plant Physiol., 2009, 149, 863–873 CrossRef CAS PubMed.
- S. D. S. Chiwocha, K. W. Dixon, G. R. Flematti, E. L. Ghisalberti, D. J. Merritt, D. C. Nelson, J.-A. M. Riseborough, S. M. Smith and J. C. Stevens, Plant Sci., 2009, 177, 252–256 CrossRef CAS.
- K. W. Dixon, D. J. Merritt, G. R. Flematti, E. L. Ghisalberti, M. Johnston, M. J. O. Dragovic and R. A. Criley, Acta Hortic., 2009, 813, 155–170 CrossRef CAS.
- M. E. Light, M. I. Daws and J. V. Staden, S. Afr. J. Bot., 2009, 75, 1–7 CrossRef CAS.
- M. G. Kulkarni, M. E. Light and J. V. Staden, S. Afr. J. Bot., 2011, 77, 972–979 CrossRef.
- G. R. Flematti, K. W. Dixon and S. M. Smith, BMC Biol., 2015, 13, 108 CrossRef PubMed.
- T. O. Sunmonu, M. G. Kulkarni and J. Van Staden, S. Afr. J. Bot., 2016, 102, 4–11 CrossRef CAS.
- J. P. Stanga, N. Morffy and D. C. Nelson, Planta, 2016, 243, 1397–1406 CrossRef CAS PubMed.
- Y. N. Yin, Acta Microbiol. Sin., 2005, 45, 837–841 CAS.
- C. S. Nautiyal, P. S. Chauhan and Y. L. Nene, J. Ethnopharmacol., 2007, 114, 446–451 CrossRef PubMed.
- A. Mandabi, H. Ganin, P. Krief, J. Rayo and M. M. Meijler, Chem. Commun., 2014, 50, 5322–5325 RSC.
- A. Delago, A. Mandabi and M. M. Meijler, Isr. J. Chem., 2016, 56, 310–320 CrossRef CAS.
- G. R. Flematti, A. Scaffidi, E. D. Goddard-Borger, C. H. Heath, D. C. Nelson, L. E. Commander, R. V. Stick, K. W. Dixon, S. M. Smith and E. L. Ghisalberti, J. Agric. Food Chem., 2010, 58, 8612–8617 CrossRef CAS PubMed.
- G. R. Flematti, E. D. Goddard-Borger, D. J. Merritt, E. L. Ghisalberti, K. W. Dixon and R. D. Trengove, J. Agric. Food Chem., 2007, 55, 2189–2194 CrossRef CAS PubMed.
- G. R. Flematti, E. L. Ghisalberti, K. W. Dixon and R. D. Trengove, J. Agric. Food Chem., 2009, 57, 9475–9480 CrossRef CAS PubMed.
- G. R. Flematti, E. L. Ghisalberti, K. W. Dixon and R. D. Trengove, Tetrahedron Lett., 2005, 46, 5719–5721 CrossRef CAS.
- E. D. Goddard-Borger, E. L. Ghisalberti and R. V. Stick, Eur. J. Org. Chem., 2007, 3925–3934 CrossRef CAS.
- K. Sun, Y. Chen, T. Wagerle, D. Linnstaedt, M. Currie, P. Chmura, Y. Song and M. Xu, Tetrahedron Lett., 2008, 49, 2922–2925 CrossRef CAS.
- R. Nagase, M. Katayama, H. Mura, N. Matsuo and Y. Tanabe, Tetrahedron Lett., 2008, 49, 4509–4512 CrossRef CAS.
- K. Matsuo and M. Shindo, Tetrahedron, 2011, 67, 971–975 CrossRef CAS.
- X.-L. Zhu, L. Xiong, H. Li, X.-Y. Song, J.-J. Liu and G.-F. Yang, ChemMedChem, 2014, 9, 1512–1521 CrossRef CAS PubMed.
- B. Helferich and M. Burgdorf, Tetrahedron, 1958, 3, 274–278 CrossRef CAS.
- W. Sowa, Can. J. Chem., 1968, 46, 1586–1589 CrossRef CAS.
- L. Xiong, X. L. Zhu, Y. Q. Shen, W. Wishwa, K. Li and G. F. Yang, Eur. J. Med. Chem., 2015, 95, 424–434 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra19424a |
| This journal is © The Royal Society of Chemistry 2016 |