
Cellulose-based spreadable new thixo gels: synthesis and their characterization†
Naresh D. Sanandiya*a and
A. K. Siddhanta*ab
aMarine Biotechnology and Ecology Discipline, CSIR-Central Salt & Marine Chemicals Research Institute, G B Marg, Bhavnagar-364002, Gujarat, India. E-mail: aks@csmcri.org; nareshsanandiya@gmail.com; Fax: +91-278-2567562; Tel: +91-278-2567760
bAcademy of Scientific & Innovative Research, Anusandhan Bhavan, 2 Rafi Marg, New Delhi-110001, India
First published on 21st September 2016
Abstract
Carboxymethyl cellulose (CMC) based new thixotropic gels were synthesized by a facile microwave-induced reaction of aminobenzoic acids (ABAs) e.g. para- and meta-aminobenzoic acids. Cellulose of the halophytic plant Salicornia brachiata was employed in this investigation. These derivatives were characterized by FT-IR, 13C NMR spectra, thermal analyses and surface morphology studies. These compounds form thixotropic soft gels in water, which were evaluated by rheological measurements. Their antioxidant properties were estimated by in vitro DPPH radical scavenging assay. These CMC-based new materials would be of potential utility in spreadable formulations in personal care applications.
Introduction
Derivatization of biopolymers is a useful approach for imparting new functional properties by grafting small active molecules onto the biopolymer backbones. The advantage of this approach is that the new material generated would possess the merits of both grafted molecules and the biopolymer aiding specific applications.Cellulose is one of the oldest highly functionalized biopolymers containing β-(1-4)-linked anhydroglucose chains, which has been receiving considerable attention for its application in food, pharmaceutical, health & personal care, and bio-medical industries. Cellulose is a biologically degradable, abundant and regenerative raw material. However, due to the inter- and intra-molecular hydrogen bonds in the cellulose structure, it neither melts nor gets dissolved readily in common solvents.1 Therefore, cellulose has been functionally modified for value addition and various applications. Cellulose can be modified by chemical reactions in many ways e.g. conversion of cellulose to carboxymethyl cellulose (CMC) is a case in point.2 CMC is a linear, long-chain, water-soluble, anionic, modified polysaccharide, which is widely used as a viscosity modulator, water binder, extrusion aid and film former in food, biomedical and cosmetics industries for providing superior consistency and flow properties to the products.2,3 Many reports are available on the synthesis and characterization of CMC, employing cellulose from different sources for example, banana rachis, sugar beet pulp, cotton stalk and rice straw, exhibiting different flow properties.4–7 Profiling of cellulose Salicornia brachiata (Sb), a halophytic plant species abundantly available along the Gujarat coast of the Arabian Sea, was reported to afford α-cellulose in high yields.8 There are no reports of employment this cellulose for preparation of new materials.
Aminobenzoic acids (ABAs) are widely distributed substance in nature, which are multifunctional hydrogen bonding molecules generating considerable interest for their biological importance and chemical properties. One of them is PABA (p-aminobenzoic acid) and its derivatives, which is used as an active ingredient in sunscreen formulation since 1943 due to its high absorbance in the UVB region (280–320 nm).9 They were frequently reported as a structural molecule in skin treatment remedies with a wide range of therapeutic applications such as treating antibacterial, antifibrotic, scleroderma, dermatomyositis10 conditions. It is used as a protective agent against photo carcinogenesis11 as well as for the treatment of Peyronie's disease.12 Few ABAs conjugated polymers/biopolymers have been reported in the literature for tissue engineering applications as wound dressing,13 colon-specific anticancer drug delivery agent,14 viscosity modulator15 as well as for preparing ion-exchange membranes.16 However, the rheological and antioxidant properties of their cellulosic derivatives have not been reported. Many aromatic compounds containing π-aromatic systems exhibited UV absorption ability, which would have potential utility as the sunscreen formulations. This is especially due to the advantages of the π-aromatic systems with an electron-donor and an electron acceptor group in the para-position.17 meta-Aminobenzoic acid (MABA) has recently been reported to exhibit interesting biological properties e.g. in facilitating cation transports in vitro and insulin secretion in mouse assays by being present in acyclic octapeptides18 and in preptin hormone,19 respectively.
Thixotropic materials tend to show lower viscosity with increasing mechanical stress and they gradually recover their original flow behavior when the applied stress is withdrawn. These materials are very important for preparing various formulations in pharmaceutical and healthcare industries.20 CMC shows a shear-thinning behavior and it is possible to modulate its rheological property by conjugating it with different substrates.
We report herein carboxymethylation of cellulose of the plant Salicornia brachiata (Sb) using microwave-mediated method followed by amidation of carboxymethyl cellulose with p-amino benzoic acid (PABA) and m-amino benzoic acid (MABA) in presence of carbodiimide coupling agent. The resulting CMC derivatives were soft thixo gels having antioxidant activity, which may be of potential utility in personal care applications.
Materials and methods
Materials
α-Cellulose, used for carboxymethylation was isolated from the stem of Salicornia brachiata collected from Diu (20° 43′56′′ N, 70° 55′45′′ E) on the west coast of India.8 A milestone Start-S Microwave lab-station for synthesis (Italy); terminal T260; line voltage 230 V; magnetron S.N. 131528; frequency 50 Hz was used to carry out the microwave induced carboxymethylation of cellulose. All chemicals were used as received, without further purification (ESI1†).Microwave-assisted carboxymethylation of cellulose
Microwave induced process has been carried out to synthesize carboxymethyl cellulose from α-cellulose of the stem of Salicornia brachiata by etherification using monochloroacetic acid and sodium hydroxide.21 The α-cellulose was converted to CMC in two steps, alkalization and etherification of cellulose under heterogeneous conditions (Fig. 1; step1). The detailed experimental procedure for CMC synthesis is furnished in the ESI1.†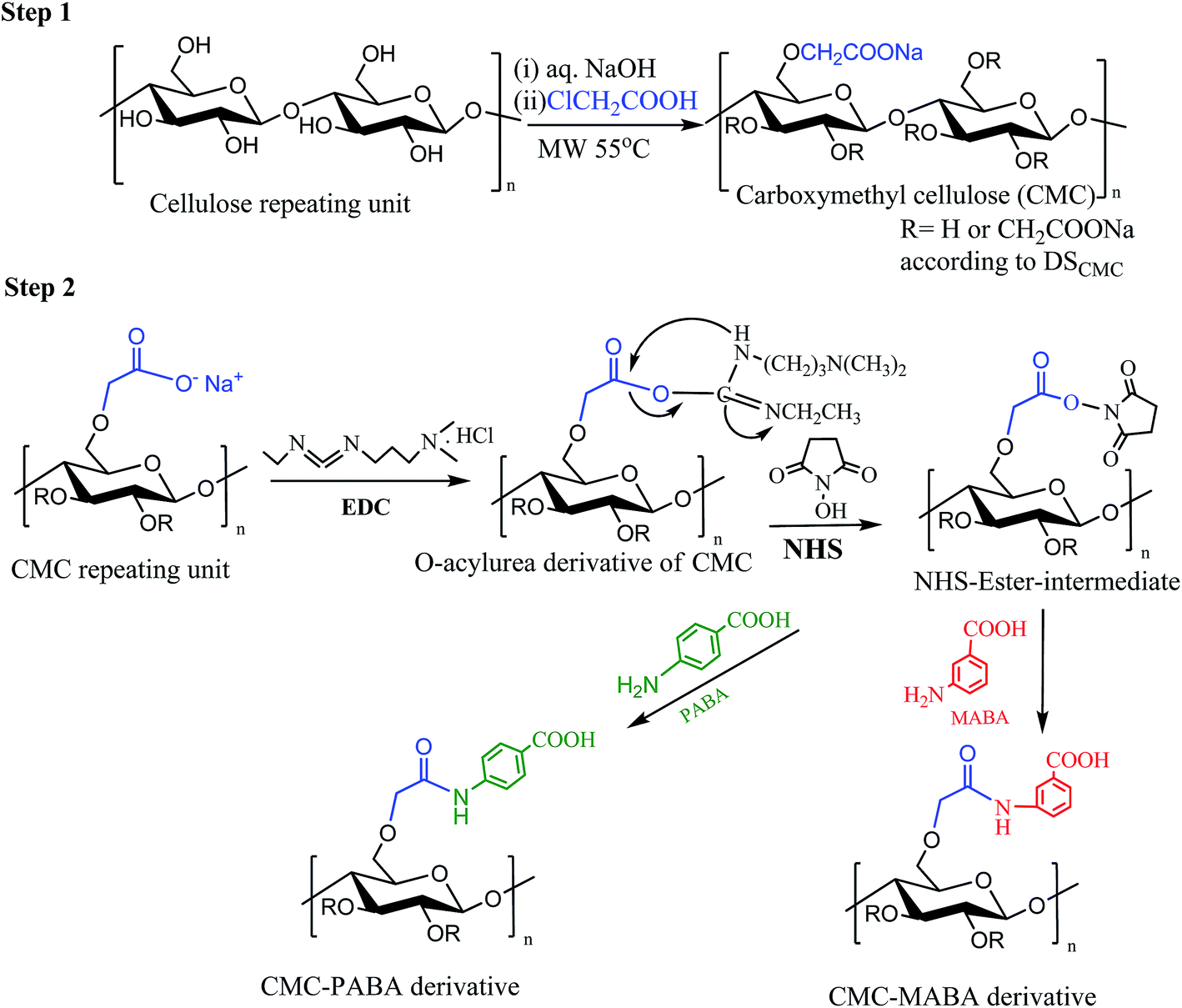 | ||
| Fig. 1 Plausible mechanism of (step 1) carboxymethylation of cellulose and (step 2) formation of CMC amino benzoic acid conjugate. | ||
The degree of substitution for CMC was determined by the standard method (ASTM, 2005).22
Synthesis of CMC–aminobenzoic acids conjugate
ABAs conjugations on the CMC backboned were carried out by carbodiimide catalyzed amidation reaction following the method reported in the literature15 (Fig. 1; step 2; ESI1†).Characterizations
CMC and CMC–aminobenzoic acid derivatives were characterized by various analytical tools e.g. 13C NMR, size exclusion chromatography (SEC), UV-visible spectroscopy, thermogravimetric analysis (TGA), differential scanning calorimetry (DSC), X-ray diffraction (XRD) and scanning electron microscopy (SEM). The detailed experimental conditions are given in the ESI2.†Rheological studies
| Ar = (Aup − Adown/Aup) × 100 | (1) |
Antioxidant activity assays
| DPPH radical scavenging activity (%) = [(A0 − A1)/A0] × 100 | (2) |
Results and discussion
The formation of CMC took place in two steps – the first step was the alkalization of the cellulose with sodium hydroxide solution (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3.24 mole equivalents) at room temperature to dismantle the bound cellulose chains, allowing water molecules to enter. In the next step, microwave-mediated etherification took place by monochloroacetic acid (1:2.05 mole equivalents) on the alkaline cellulose chains. The amide conjugate of CMC and aminobenzoic acid was catalyzed by carbodiimide through activation of the carboxylate functionality. Carbodiimide (EDC) reacted with carboxyl groups to form an unstable, intermediate O-acylurea, followed by activation by N-hydroxysuccinimide (NHS). The NHS-ester intermediate thus formed reacted with the primary amine groups to form amide derivative. ortho-Amino benzoic acid did not give rise to the expected amide derivative with CMC. Chhatbar et al. (2012) reported, however, that p-amino benzoic acid did not form amide with sodium alginate citing the difference in the pKa values of aminobenzoic acids vis a vis alginic acid.15 The pKa values of o-, m- and p-aminobenzoic acids are 6.97, 4.78 and 4.92 respectively.25 The pKa values of alginic acid are 3.38 & 3.65 (ref. 26) and that of CMC is 3.2.27 It appears that the relative differences in the pKa values of amino benzoic acids and CMC when compared with those of alginic acid, may be playing a crucial role in the selectivity of the amide formation with para-aminobenzoic acid as opposed to ortho-aminobenzoic acid.
:3.24 mole equivalents) at room temperature to dismantle the bound cellulose chains, allowing water molecules to enter. In the next step, microwave-mediated etherification took place by monochloroacetic acid (1:2.05 mole equivalents) on the alkaline cellulose chains. The amide conjugate of CMC and aminobenzoic acid was catalyzed by carbodiimide through activation of the carboxylate functionality. Carbodiimide (EDC) reacted with carboxyl groups to form an unstable, intermediate O-acylurea, followed by activation by N-hydroxysuccinimide (NHS). The NHS-ester intermediate thus formed reacted with the primary amine groups to form amide derivative. ortho-Amino benzoic acid did not give rise to the expected amide derivative with CMC. Chhatbar et al. (2012) reported, however, that p-amino benzoic acid did not form amide with sodium alginate citing the difference in the pKa values of aminobenzoic acids vis a vis alginic acid.15 The pKa values of o-, m- and p-aminobenzoic acids are 6.97, 4.78 and 4.92 respectively.25 The pKa values of alginic acid are 3.38 & 3.65 (ref. 26) and that of CMC is 3.2.27 It appears that the relative differences in the pKa values of amino benzoic acids and CMC when compared with those of alginic acid, may be playing a crucial role in the selectivity of the amide formation with para-aminobenzoic acid as opposed to ortho-aminobenzoic acid.
The CMC derivatives synthesized with MABA and PABA under optimum reaction conditions were used for characterizations. Primary amino (–NH2) groups were not detected in the CMC–MABA and CMC–PABA as well as in the non-modified CMC samples using the TNBS-reagent method indicating complete removal of the unreacted MABA and PABA from the reaction mixture during the dialysis process. Total nitrogen contents of CMC, CMC–MABA and CMC–PABA were determined and are presented in Table 1.
| Samples | % yielda (±SD) | N% (±SD) | Apparent viscosityb (cP) | Mw (kDa) | PDI |
|---|---|---|---|---|---|
| a Data presented are mean of triplicate measurements (±SD).b Apparent viscosity measured at conc. (1% w/v) at 30 rpm using Spindle SC4-18 and at 25 °C on a Brookfield viscometer; NA-not applicable. | |||||
| CMC | 91.50 ± 0.5 | NA | 3.20 ± 0.20 | 132 | 2.25 |
| CMC–MABA | 71.56 ± 0.5 | 1.81 ± 0.02 | 18.60 ± 0.20 | 170 | 5.29 |
| CMC–PABA | 67.03 ± 0.5 | 1.49 ± 0.02 | 9.20 ± 0.20 | 156 | 3.22 |
Optimization study revealed that CMC with 91.5% yield and 1.07 DS was obtained with 1:3.24:2.05 (AGU:NaOH:MCA) mole equivalents with respect to AGU (anhydroglucose units), whereas CMC–MABA and CMC–PABA with 1:1 mole equivalent with respect to CMC, led to the formation of the amide conjugate in maximum yield and DS. The yields of CMC–MABA and CMC–PABA were 71.56% and 67.03% having nitrogen contents 1.81% and 1.49%, respectively (Table 1). The respective degrees of substitution in CMC–MABA and CMC–PABA were 0.44 and 0.36. It may be noted that the CMC derivatives synthesized employing MABA and PABA were soluble in water under ambient conditions. CMC (1% w/v) showed apparent viscosities of 3.20 ± 0.20 cP at 25 °C, the apparent viscosities measured for the derivatives under identical experimental conditions showed much higher values, which were 18.60 ± 0.20 cP and 9.20 ± 0.20 cP for CMC–MABA and CMC–PABA derivatives, respectively (Table 1). The increase in viscosities may be due to the formation of the stronger double helical structures through the participation of –COOH group of MABA and PABA in the formation of hydrogen bonding subsequently increasing the number of junction zones in the gel network system.15 Additionally, in protic solvents, ABAs act both as H acceptors at the O atom of the carboxylic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group & at the N atom of the –NH2 group as well as act as H donors at the –OH group & at the –NH2 group and can also form hydrogen bonds through their aromatic π-electrons.28 The weight-average molecular weights (Mw) of CMC–MABA and CMC–PABA were 170 kDa (Mw/Mn = 5.29) and 156 kDa (Mw/Mn = 3.22) respectively. This value represents an increase from the initial Mw of CMC (Mw) 132 kDa, (Mw/Mn = 2.25) (Table 1). Thus the enhancement of viscosity may be attributed to the upstream departure of Mw/Mn (polydispersity index, PDI) from unity in the products reflecting their non-linear geometry29 coupled with the facilitation of hydrogen bonding through the participation of the carboxyl groups.
O group & at the N atom of the –NH2 group as well as act as H donors at the –OH group & at the –NH2 group and can also form hydrogen bonds through their aromatic π-electrons.28 The weight-average molecular weights (Mw) of CMC–MABA and CMC–PABA were 170 kDa (Mw/Mn = 5.29) and 156 kDa (Mw/Mn = 3.22) respectively. This value represents an increase from the initial Mw of CMC (Mw) 132 kDa, (Mw/Mn = 2.25) (Table 1). Thus the enhancement of viscosity may be attributed to the upstream departure of Mw/Mn (polydispersity index, PDI) from unity in the products reflecting their non-linear geometry29 coupled with the facilitation of hydrogen bonding through the participation of the carboxyl groups.
Characteristic IR bands of Sb-cellulose were (Fig. 2a): νmax (cm−1) 3417 br (–OH stretching vibration), 2902 w (CH stretching vibration), 1643 s H–O–H polymer bound water), 1433 w (C–C bending), 1059 s br (C–O–C stretching vibration), 950–680 cm−1 (the fingerprint or anomeric region of carbohydrates). Whereas, FTIR spectra of CMC (Fig. 2b), absorption bands appeared at 1606 cm−1 (stretching vibration of COO−), 1421 (–CH scissoring vibrations 1324 cm−1 (–OH) bending vibrations 2918 cm−1 (stretching vibration –C–H) confirmed the etherification.27 The absorption band of the carbonyl (CO) stretching of the secondary amide at 1547 cm−1 and 1540 cm−1 and a medium-sharp peak at 1266 and 1260 cm−1 (amide II band, interaction between the N–H bending vibrations) of CMC–MABA and CMC–PABA, respectively confirmed the amide bond formation (Fig. 2c and d).
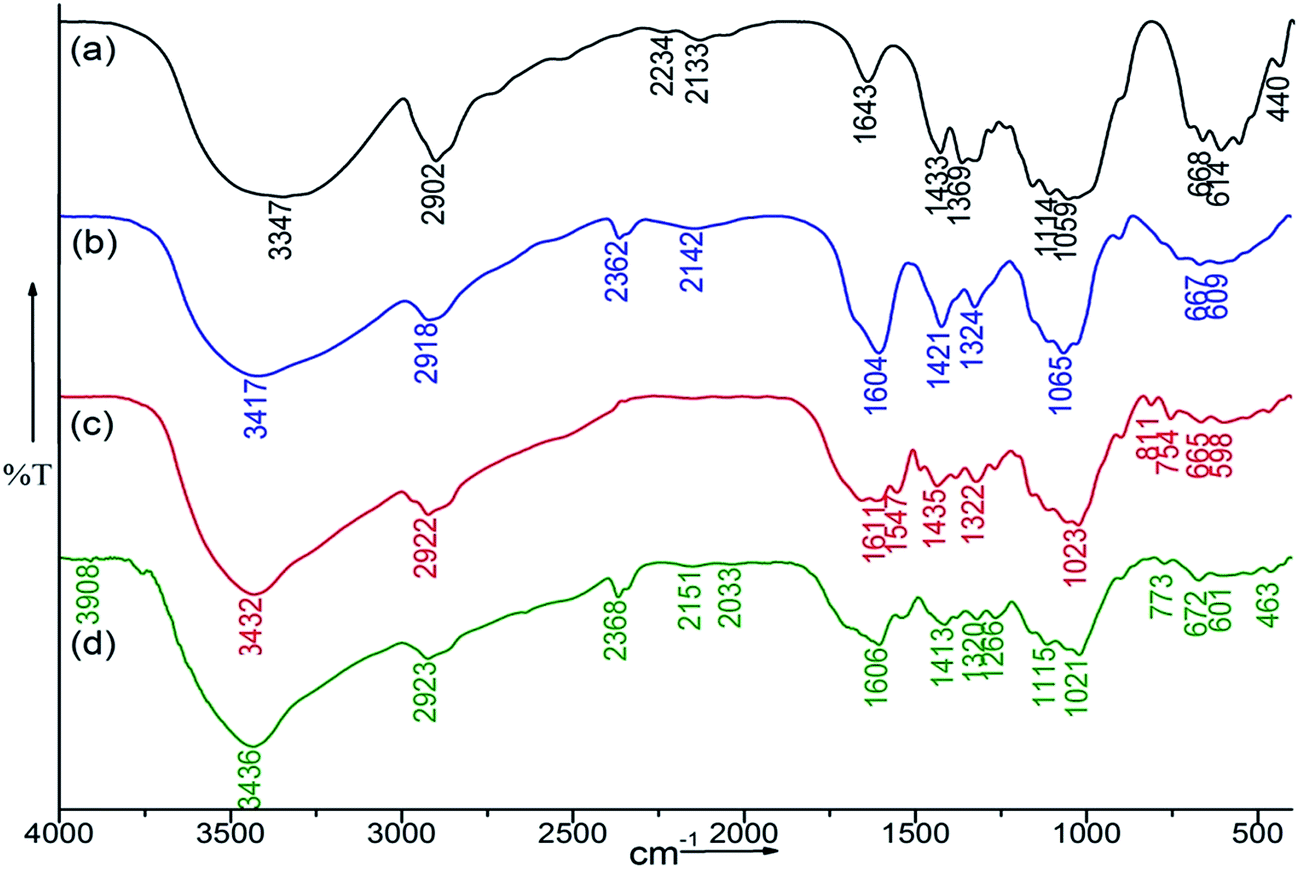 | ||
| Fig. 2 FTIR spectra of (a) Sb-cellulose, (b) CMC, (c) CMC–MABA and (d) CMC–PABA. | ||
The 13C NMR resonances of CMC, CMC–MABA, CMC–PABA, MABA and PABA with their probable assignment of the δ values are given in Table 2, Fig. 3 and S1.† The assignments were done by comparison with the corresponding data reported in the literature (Ramos et al., 2005; cf. Chhatbar et al., 2012) as well as with those obtained by the ChemDraw (v11.0) structures.15,27
| Assignments | CMC | MABA | CMC–MABA (δ ppm) | PABA | CMC–PABA |
|---|---|---|---|---|---|
| C-1 | 103.9 | — | 103.9 | — | 104.1 |
| C-2 | 74.6 | — | 74.6 | — | 74.8 |
| C-2s | 75.6 | 75.8 | 759 | ||
| C-3 | 72.8 | — | 72.5 | — | 73.0 |
| C-4 | 79.8 | — | 80.2 | — | 80.4 |
| C-5 | 76.4 | — | 76.5 | — | 76.6 |
| C-6 | 61.4 | — | 61.7 | — | 62.0 |
| C-7 | 71.8 | — | 72.1 | — | 71.5 |
| C-8 | 179.3 | — | 178.7 | — | 178.6 |
| C-1′ | — | 129.0 | 127.7 | 117.0 | 130.5 |
| C-2′ | — | 114.6 | 124.3 | 131.4 | 132.3 |
| C-3′ | — | 148.9 | 130.7 | 112.7 | 122.6 |
| C-4′ | — | 116.9 | 127.0 | 153.2 | 141.8 |
| C-5′ | — | 131.4 | 127.7 | 112.7 | 122.6 |
| C-6′ | — | 118.2 | 127.0 | 131.4 | 132.3 |
| C-7′ | — | 168.1 | 172.7 | 167.7 | 172.5 |
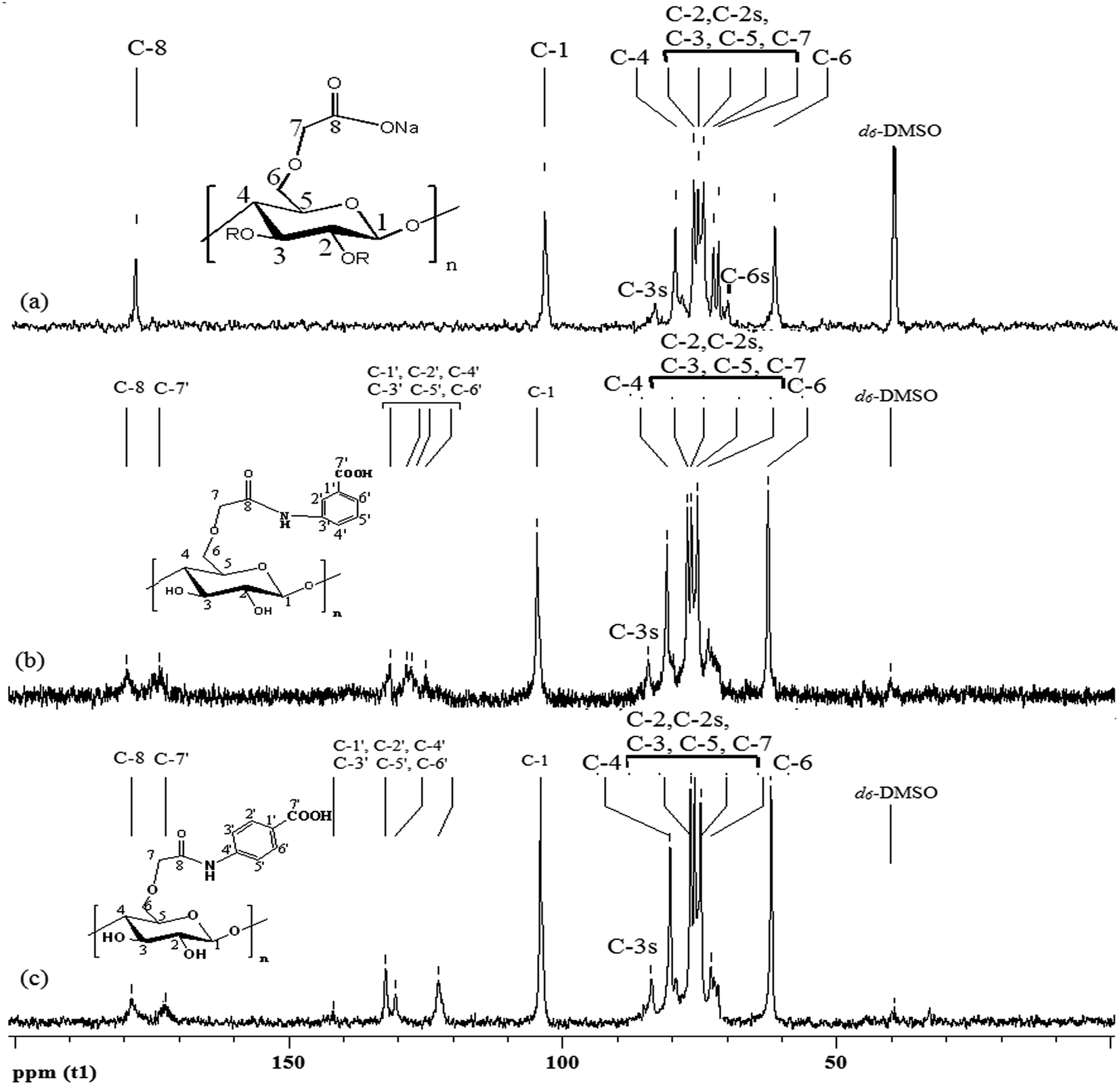 | ||
| Fig. 3 13C NMR spectra of (a) CMC, (b) CMC–MABA, (c) CMC–PABA. The index ‘s’ refers to substituted hydroxyl group in CMC. | ||
In CMC, the carbon resonance peak at 178.3 ppm was attributed to the –C(8)OOH group resulted from the carboxymethylation of cellulose and peaks in the range 61.4–103.9 ppm were attributed to the C-1 to C-7 of CMC, which confirmed the carboxymethylation of cellulose (Fig. 3a). Seven carbons of MABA (C′-1, C′-3, C′-5, C′-6, C′-4, C′-2 and C′-7) appeared at 129.0, 114.6, 148.9, 116.9, 131.4, 118.2 and 168.1, and those of PABA appeared at 117.0, 131.4, 112.7, 153.2, 112.7, 131.4 and 167.7 respectively (ESI1†). The 13C NMR resonances of CMC–MABA, the new peaks at 127.7 (C′-1, C′-5), 124.3 (C′-2), 130.7 (C′-3), 127.0 (C′-4, C′-6) and 172.7 (C′-7) were assigned to the carbons of MABA moieties. In CMC–PABA, the new peaks at 130.5 (C-1′), 132.3 (C-2′, C-6′), 122.6 (C-3′, C-5′), 141.8 (C-4′), 172.5 (C-7′) were assigned to PABA moieties (Fig. 3). In addition, shifting of the carbonyl peaks at 178.7 and 178.6 ppm in CMC–MABA and CMC–PABA, and an upfield shift of C-3′ of MABA and C-4′ of PABA in the amide derivatives confirmed the amidation (Table 2; Fig. 3b and c).
The absorption maxima of MABA (10−3 M in water) in UV spectra appeared at λmax 208 nm and 300 nm whereas; PABA at λmax 266 nm (ESI2†). However, CMC did not have any absorption in the UV-vis range. The amide conjugate CMC–MABA (1 × 10−2 M) exhibited UV absorption band at λmax at 209 nm and 298 nm, while in CMC–PABA the absorption band appeared at 262 nm (Fig. S2†), indicating amide formation.
SEM images of Sb-cellulose, CMC, CMC–MABA and CMC–PABA exhibited distinctly different surface morphology, which indicated significant changes on the surface morphologies (Fig. 4). Sb-Cellulose appeared to exhibit discrete fibrous morphology indicating cellulosic surface, whereas the surface morphology of CMC appeared to have a similar fibrous structure having more compact and organized arrangement. The modified CMC–MABA and CMC–PABA derivatives lost the fibrous morphology and exhibited even more compact and porous structure, clearly indicating modification.
 | ||
| Fig. 4 SEM images of (a) Sb-cellulose, (b) CMC, (c) CMC–MABA and (d) CMC–PABA [scale bar: 10 μm]. | ||
The thermal behavior of CMC, CMC–MABA and CMC–PABA are shown in the thermograms (Fig. S3†). The thermal stability profile was CMC > CMC–MABA > CMC–PABA. The thermogram of CMC exhibited first mass loss (ca. 5%) in the temperature range 30–250 °C followed by sharp mass losses (ca. 32%) in the range 250–300 °C and further, with gradual mass losses till 750 °C leaving behind ∼47% residue. Similar steps in the mass loss were observed in CMC–MABA and CMC–PABA. In the range 30–250 °C ca. 15.0% mass loss occurred which may be attributed to the loss of the polymer bound water. This was followed by a sharp mass loss (totalling ca. 50%) in the range 250–350 °C and, and a gradual mass loss up to (totalling ca. 70%) due to the decomposition of the polymer backbone in the range 350–750 °C. The thermal stability changed in the modified derivative than that in the parent polymer (CMC), which further corroborated modification on the polymer backbone.
The DSC thermograms (−20 °C to 25 °C@2 °C min−1) of the CMC, CMC–MABA and CMC–PABA are presented in Fig. 5A. They showed Tg values: CMC (Tg −5.95 °C), CMC–MABA (Tg −7.17 °C), CMC–PABA (Tg −6.02 °C), exhibiting their phase transitions. The DSC thermograms (30–500 °C, @10 °C min−1) of the CMC, CMC–MABA and CMC–PABA are presented in Fig. 5B. The thermograms of CMC showed broad endothermic peaks at a 139.5 °C and two exothermic peaks at 295 °C and 330 °C whereas, in CMC–MABA and CMC–PABA derivatives, endothermic and exothermic peaks appeared at 103.5 °C and 283 °C, respectively. The difference in temperatures may be attributed to the difference in their respective hydrophilicity.30
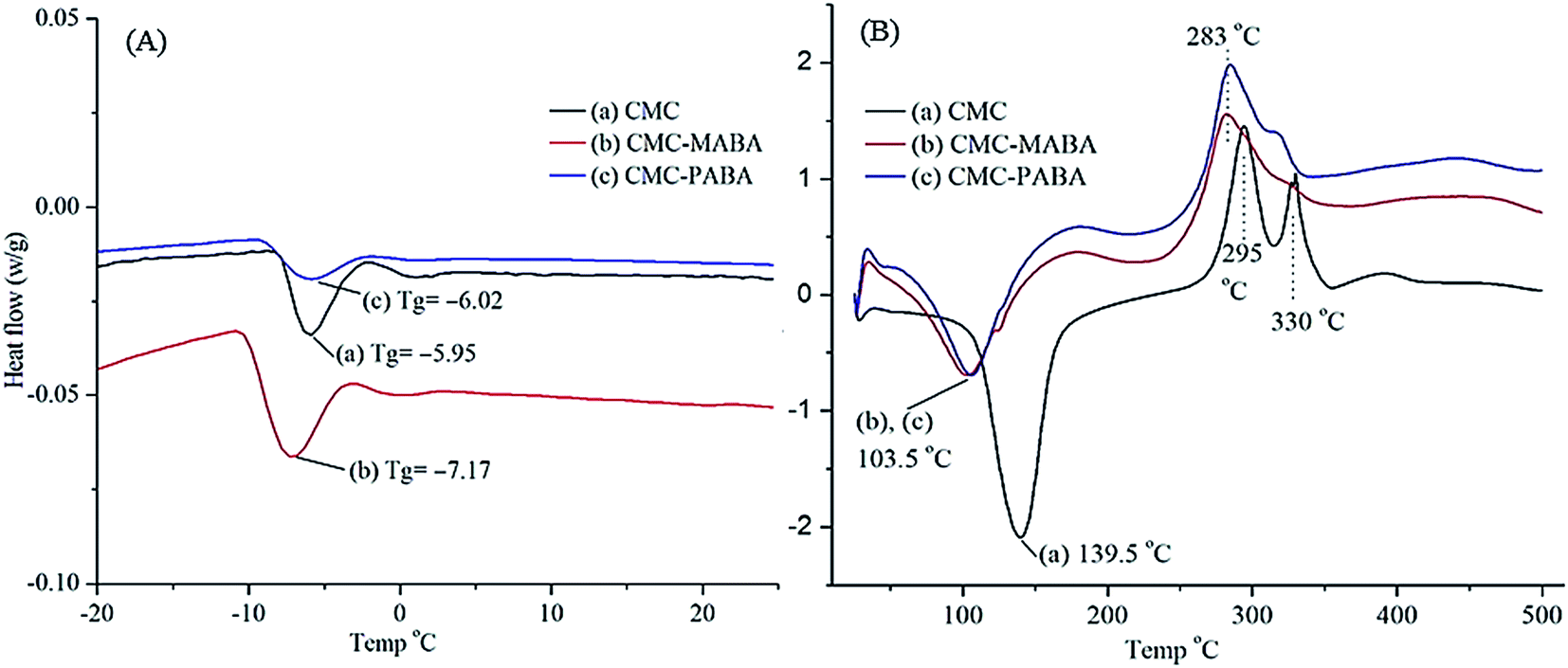 | ||
| Fig. 5 Differential scanning calorimetry of (a) CMC, (b) CMC–MABA and CMC–PABA at temperature range (A) −20 °C to +25 °C; heating rate@2 °C min−1 and (B) +25 °C to +550 °C; heating rate 10 °C min−1. | ||
The X-ray diffraction patterns of cellulose, CMC, MABA, CMC–MABA, PABA and CMC–PABA are depicted in Fig. 6. The X-ray diffraction patterns of CMC indicated its amorphous structure as against cellulose, which exhibited crystalline structure (peaks at 2θ = 12° and a twin peak at 2θ = 20°). MABA exhibited several sharp peaks and PABA exhibited five sharp peaks, indicating their crystalline nature. The XRD patterns of CMC–MABA and CMC–PABA also showed many sharp peaks of respective amino benzoic acid residues, suggesting induction of crystallinity on the backbone of CMC.
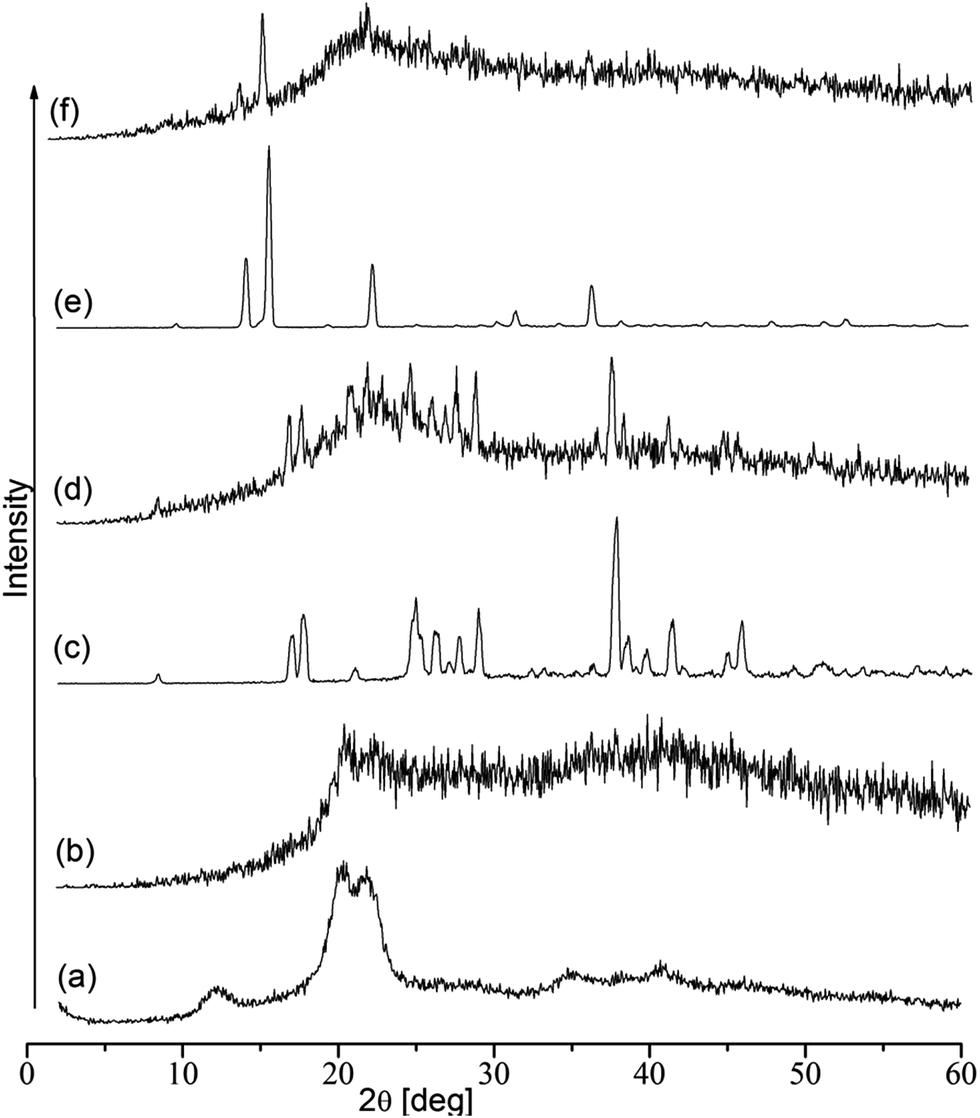 | ||
| Fig. 6 X-ray diffraction patterns of (a) cellulose, (b) CMC, (c) MABA, (d) CMC–MABA, (e) PABA and (f) CMC–PABA. | ||
The shear viscosity data of the CMC, CMC–MABA and CMC–PABA at the concentration 4% are depicted in Fig. S4.† The dynamic viscosity (η) decreased with increasing shear rate (γ), corresponding to a shear thinning behavior for all the samples, indicating the shear thinning behavior of the samples.
In time-dependent thixotropic behavior experiment, CMC–MABA and CMC–PABA showed good thixotropic behavior than CMC at a concentration 4.0%, which exhibited variations in the viscosities with the time at the applied shear rate in three different steps (Fig. 7a). In the first step, viscosity (3.87 to 3.77 Pa s) of CMC–MABA gradually decreased with time at a shear rate 1 s−1. In the second step, on applying shear rate 100 s−1 there was an immediate drop in the viscosity (3.77 to 0.53 Pa s) with time. In the third step on the application of low shear rate (changed from 100 s−1 to 1 s−1), the compound showed an increment in the viscosity (0.41 to 1.82 Pa s) with time, exhibiting a time-dependent thixotropic behavior of the CMC–MABA. However, in CMC–PABA, a reverse trend was observed in the first step, which implied increment in viscosity (1.37 to 1.55 Pa s). In the second step, an immediate fall in viscosity was observed (1.55 to 0.19 Pa s) with time. In the third step, the compound showed an increase in viscosity (0.15 to 0.82) with time exhibiting the time-dependent thixotropic behavior of CMC–PABA. CMC, however, exhibited lowest time-dependent thixotropic behavior than CMC–MABA and CMC–PABA (Fig. 7a). Therefore, it can be concluded that introduction of MABA and PABA improved thixotropicity on CMC, marginally though, possibly due to the change in the molecular arrangements in the amide derivatives. It may be noted here that greater thixotropicity in amide conjugate arose perhaps due to the relative increase in hydrophobicity inhibiting thereby the formation of an interpenetrating networks. These observations tend to suggest that the presence of favorable hydrophilic–lipophilic balance (HLB) must be playing a crucial role for the thixotropic characteristics.
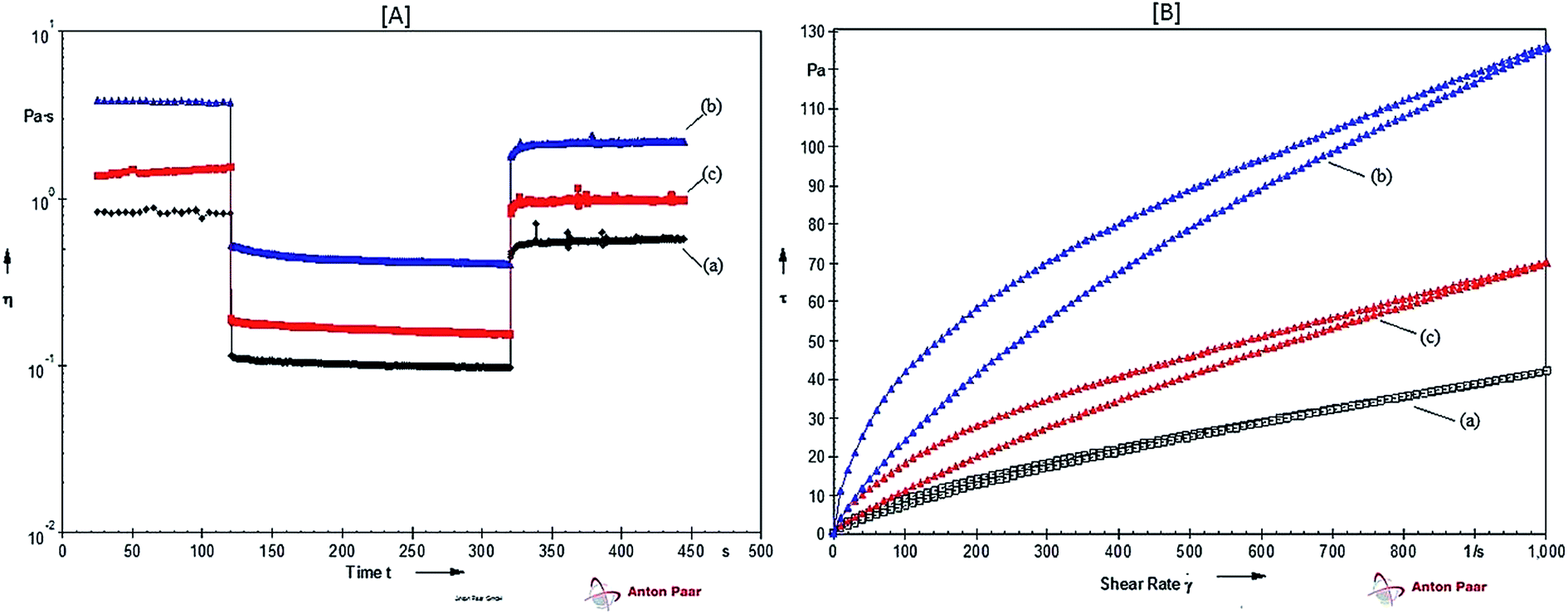 | ||
| Fig. 7 [A] Time-dependent thixotropicity and [B] evaluation of thixotropic behavior by hysteresis loop measurements of (a) CMC, (b) CMC–MABA and (c) CMC–PABA at 4% w/v concentration. | ||
For the evaluation of thixotropic behavior by hysteresis loop test, the samples were subjected to thinning under increasing shear rate (upward curve) immediately followed by dropping the applied shear rate (downward curve) (Fig. 7b). The area of the loop thus formed gave an idea of the extent of thixotropicity present in the samples, which was calculated using eqn (1). The relative percentage of thixotropy area (Ar) of CMC–MABA and CMC–PABA were 11.40% and 10.16%, respectively, which were greater than those of CMC (2.79%) indicating relatively superior thixotropy in both the amide conjugates to that of CMC at a concentration 4.0%. The enhanced thixotropicity of CMC–ABAs as against that of CMC may presumably be due to the difference in structures of the guest molecules leading to the altered molecular arrangements inducing thereby additional Intermolecular hydrogen bonding interactions stabilizing the supramolecular systems.31
DPPH is a stable radical and widely used to evaluate antioxidant activities of substances. The antioxidant activity of CMC and its derivatives were carried out by the measurement of absorbance of DPPH radical at 517 nm in comparison to that of control. As shown in Fig. 8, it was observed that the DPPH free radical scavenging abilities of CMC–MABA and CMC–PABA derivatives were concentration dependent, albeit less potent than the standard BHT (85% at conc. 30 μg ml−1) showing respective activities a maximum ca. 37.41% (concentration; 160 μg ml−1) and 55.0% (concentration; 200 μg ml−1). The EC50 of BHT and CMC–PABA were 11.46 μg ml−1 and 128.38 μg ml−1, respectively. However, EC50 of CMC–MABA could not be obtained in the measurement range. Control CMC sample did not show scavenging activity towards DPPH free radical. This study suggested conjugation of aminobenzoic acid moieties on the CMC backbone imparted DPPH radical scavenging activity and they could serve as free radical scavengers, acting mainly as a primary natural antioxidant.
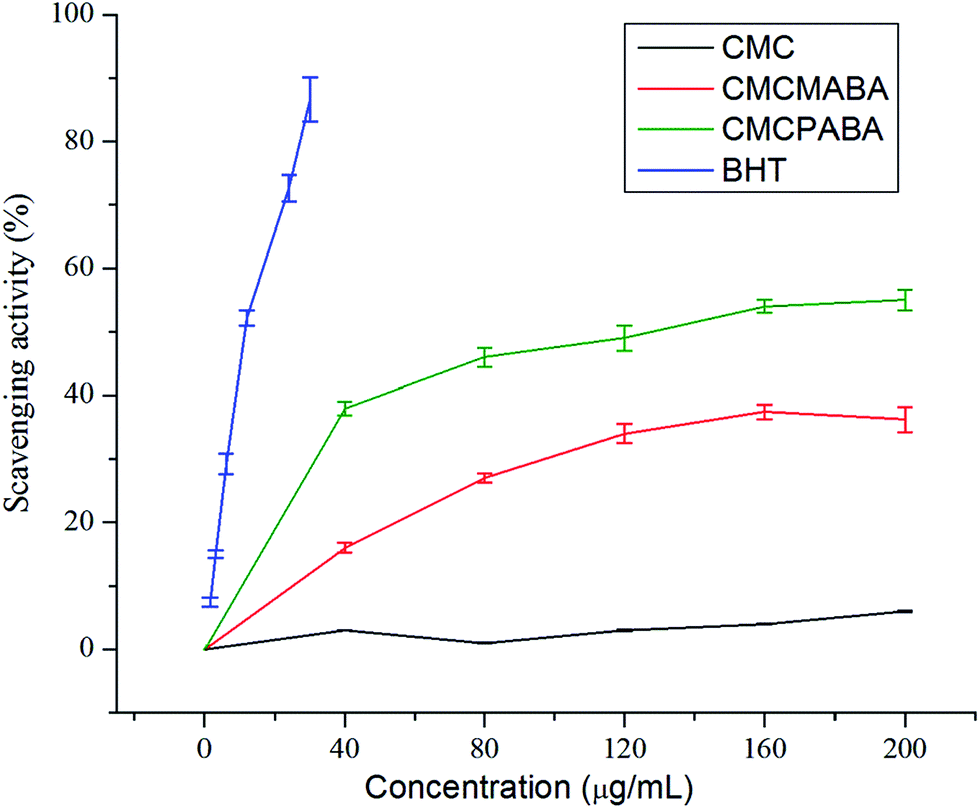 | ||
| Fig. 8 DPPH free radical scavenging assay of CMC, CMC–MABA and CMC–PABA. | ||
Conclusions
α-Cellulose of the halophyte Salicornia brachiata of the Arabian Sea coast of India was employed to synthesize its carboxymethyl derivative, which was then coupled with para-aminobenzoic (PABA) and meta-aminobenzoic (MABA) acids to produce corresponding new amide derivatives, CMC–PABA and CMC–MABA. These amides exhibited relatively superior thixotropicity to that of CMC at the same concentration. In DPPH free radical scavenging assay they showed 37.41% (concentration 160 μg ml−1) and 55.0% (concentration 200 μg ml−1) antioxidant activity respectively compared to that of standard BHT's 85% at a concentration 30 μg ml−1. These cellulose-based spreadable thixotropic polymeric compounds having antioxidant activity may be of potential utility in developing materials for applications in the domain of pharmaceutical and personal/skin care applications.Acknowledgements
CSIR-CSMCRI Communication No. 109/2016. Sincere thanks are accorded to CSIR, New Delhi, for the funding under the CSIR Emeritus Scientist Scheme awarded to AKS. Thanks are accorded to CSIR for an award of senior research fellowship to NDS as well as for the generous funding support towards infrastructure and core competency development under the Analytical Science Discipline and Centralized Instrument Facility.References
- K. Hattori, E. Abe, T. Yoshida and J. A. Cuculo, Polym. J., 2004, 36, 123–130 CrossRef CAS.
- Y. Habibi, Chem. Soc. Rev., 2014, 43, 1519–1542 RSC.
- H. N. Cheng, M. Takai and E. A. Ekong, Macromol. Symp., 1999, 140, 145–153 CrossRef CAS.
- H. Toğrul and N. Arslan, Carbohydr. Polym., 2003, 54, 73–82 CrossRef.
- M. P. Adinugraha, D. W. Marseno and Haryadi, Carbohydr. Polym., 2005, 62, 164–169 CrossRef CAS.
- F. A. Abdel-Mohdy, E. S. Abdel-Halim, Y. M. Abu-Ayana and S. M. El-Sawy, Carbohydr. Polym., 2009, 75, 44–51 CrossRef CAS.
- G. Zhang, L. Zhang, H. Deng and P. Sun, J. Chem. Technol. Biotechnol., 2011, 86, 584–589 CrossRef CAS.
- N. D. Sanandiya, K. Prasad, R. Meena and A. K. Siddhanta, Nat. Prod. Commun., 2010, 5, 603–606 CAS.
- F. P. Gasparro, M. Mitchnick and J. F. Nash, Photochem. Photobiol., 1998, 68, 243–256 CrossRef CAS PubMed.
- M. R. Griffiths and G. C. Priestley, Acta Derm.-Venereol., 1992, 72, 15–18 CAS.
- H. Flindt-Hansen, P. Thune and T. Eeg Larsen, Arch. Dermatol. Res., 1990, 282, 38–41 CrossRef CAS PubMed.
- W. Weidner, E. W. Hauck and J. Schnitker, Eur. Urol., 2005, 47, 530–536 CrossRef CAS PubMed.
- M. Gizdavic-Nikolaidis, S. Ray, J. R. Bennett, A. J. Easteal and R. P. Cooney, Macromol. Biosci., 2010, 10, 1424–1431 CrossRef CAS PubMed.
- T. Plyduang, L. Lomlim, S. Yuenyongsawad and R. Wiwattanapatapee, Eur. J. Pharm. Biopharm., 2014, 88, 351–360 CrossRef CAS PubMed.
- M. U. Chhatbar, K. Prasad, D. R. Chejara and A. K. Siddhanta, Soft Matter, 2012, 8, 1837–1844 RSC.
- A. H. Rosa, D. Goveia, I. C. Bellin, S. da Silva Lessa, N. L. Dias Filho and P. de Magalhães Padilha, Anal. Bioanal. Chem., 2006, 386, 2153–2160 CrossRef CAS PubMed.
- R. Jansen, U. Osterwalder, S. Q. Wang, M. Burnett and H. W. Lim, J. Am. Acad. Dermatol., 2013, 69, 867 CrossRef PubMed.
- B. P. Benke and N. Madhavan, Chem. Commun., 2013, 49, 7340–7342 RSC.
- C. M. Buchanan, Z. Peng, A. Cefre and V. Sarojini, Chem. Biol. Drug Des., 2013, 82, 429–437 CAS.
- J. Mewis and N. J. Wagner, Adv. Colloid Interface Sci., 2009, 147–148, 214–227 CrossRef CAS PubMed.
- J. P. Chaudhary, S. Kondaveeti, V. Gupta, K. Prasad and R. Meena, J. Appl. Polym. Sci., 2014, 131, 40630 Search PubMed.
- ASTM, CK-G06 Edition, 2005, 05, D-1439-1403.
- M. Dolz, F. González, J. Delegido, M. J. Hernández and J. Pellicer, J. Pharm. Sci., 2000, 89, 790–797 CrossRef CAS PubMed.
- G.-C. Yen and H.-Y. Chen, J. Agric. Food Chem., 1995, 43, 27–32 CrossRef CAS.
- CRC Handbook of Chemistry and Physics, ed. R. C. Weast and M. J. Astle, CRC Press Inc, Boca Raton, Florida, 1980 Search PubMed.
- I. Donati and S. Paoletti, Alginates: Biology and Applications, Springer, Berlin/Heidelberg, 2009, vol. 13, pp. 1–53 Search PubMed.
- T. Heinze and A. Koschella, Macromol. Symp., 2005, 223, 13–40 CrossRef CAS.
- Y. He, C. Wu and W. Kong, J. Phys. Chem. A, 2005, 109, 748–753 CrossRef CAS PubMed.
- K. Abu-Rabeah, B. Polyak, R. E. Ionescu, S. Cosnier and R. S. Marks, Biomacromolecules, 2005, 6, 3313–3318 CrossRef CAS PubMed.
- N. D. Sanandiya and A. K. Siddhanta, Carbohydr. Res., 2013, 381, 93–100 CrossRef CAS PubMed.
- Y. Zhang, S. Yu and F. Bao, Carbohydr. Res., 2008, 343, 2504–2508 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra19264e |
| This journal is © The Royal Society of Chemistry 2016 |