
Highly dispersed palladium nanoparticles on poly(N1,N3-dimethylbenzimidazolium)iodide-functionalized multiwalled carbon nanotubes for ethanol oxidation in alkaline solution†
Abstract
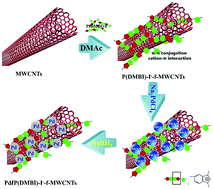
Multi-walled carbon nanotubes (MWCNTs) have been considered as good catalyst supporting materials, and their dispersion and functionalization are important, challenging problems for high-performance composite catalysts. Here, poly(N1,N3-dimethylbenzimidazolium)iodide (P(DMBI)-I−) was successfully synthesized by methylation of polybenzimidazole (PBI) for the dispersion and functionalization of MWCNTs. The results demonstrate that the novel P(DMBI)-I− exhibits a higher dispersion effect for MWCNTs than the original PBI in some typical organic solvents. MWCNTs were wrapped with a thin layer of P(DMBI)-I− and achieved functionalization with π–π conjugation and cation–π interaction. Benefiting from the positive charges and imidazole rings of P(DMBI)-I−, the palladium nanoparticles/P(DMBI)-I−-functionalized MWCNTs hybrid (Pd/P(DMBI)-I−-f-MWCNTs) catalyst loaded with highly dispersed palladium nanoparticles was fabricated by in situ reduction. TEM and SEM images demonstrated that when the feed weight ratio of Na2PdCl4 and P(DMBI)-I−-f-MWCNTs was 6 : 1, the Pd NPs of Pd/P(DMBI)-I−-f-MWCNTs with particle size of ∼2.9 nm were well distributed on P(DMBI)-I−-f-MWCNTs in good quantity. The amount of Pd loading on the catalyst of Pd/P(DMBI)-I−-f-MWCNTs was about 56.43 wt%. With respect to ethanol oxidation in alkaline solution, the Pd/P(DMBI)-I−-f-MWCNTs exhibited higher electrochemical performance and tolerance stability, compared to commercial Pd/C and Pd/PBI-f-MWCNTs.
 Please wait while we load your content...
Please wait while we load your content...

