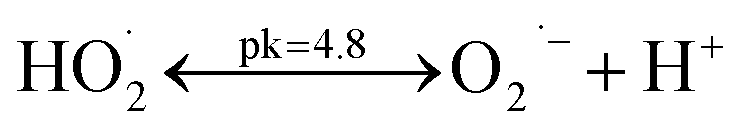DOI:
10.1039/C6RA19191F
(Paper)
RSC Adv., 2016,
6, 87569-87583
Heterogeneous photocatalytic ozonation of ciprofloxacin using synthesized titanium dioxide nanoparticles on a montmorillonite support: parametric studies, mechanistic analysis and intermediates identification†
Received
28th July 2016
, Accepted 6th September 2016
First published on 7th September 2016
Abstract
A titanium dioxide/montmorillonite (TiO2/MMT) nanocomposite was prepared as a photocatalyst by a hydrothermal method. The physicochemical properties of the prepared sample were comprehensively characterized using scanning electron microscopy (SEM), electron dispersive X-ray spectroscopy (EDX), high resolution transmission electron microscopy (HRTEM), Fourier transform infrared spectroscopy (FTIR), X-ray fluorescence (XRF), N2 adsorption–desorption, ultraviolet-visible diffuse reflectance spectroscopy (UV-vis DRS), and the pH of the zero point of charge (pHzpc) analysis. The photocatalytic ozonation of ciprofloxacin (CIP) was studied in the presence of the TiO2/MMT nanocomposite under different experimental conditions. Comparison of the main processes such as photocatalysis, ozonation and photocatalytic ozonation revealed that photocatalytic ozonation resulted in the highest degradation efficiency (90.00% at 30 min) of the pollutant under the optimum conditions ([CIP]0 = 20 mg L−1, [catalyst]0 = 0.04 g L−1, ozone gas flow rate = 2 L h−1 and pH = 5). This increase was due to a synergistic effect between photocatalysis and ozonation triggered by TiO2/MMT. The mechanism of the photocatalytic ozonation process was investigated in the presence of various organic and inorganic reactive oxygen species (ROS) scavengers. Accordingly, among the radical scavengers, the iodide ions and benzoquinone showed the highest inhibitory effect on the degradation efficiency of CIP. The photocatalytic ozonation mechanism of the TiO2/MMT nanocomposite for the degradation of CIP was thoroughly investigated. The performance of the photocatalytic ozonation process in a real water matrix was evaluated using well and ground water samples. In addition, the reusability of the TiO2/MMT nanocomposite in the photocatalytic ozonation process was examined. The result showed that the degradation efficiency of CIP declines by only about 7% after four consecutive runs. The main degradation intermediates of CIP produced in the photocatalytic ozonation process were identified by gas chromatography coupled to mass spectrometry (GC-MS) analysis.
1. Introduction
Nowadays, personal care products (PCPs) and pharmaceuticals are considered significant contributors to water contamination, due to their hazardous biological and ecotoxicological effects. According to the literature, most pharmaceuticals are extremely resistant to conventional wastewater treatment methods, so they persist within water cycles and remain in the environment.1,2 This leads to pollution and other disturbing effects.3–6 For example, ciprofloxacin (CIP) is an antibacterial pharmaceutical, which is classified as a second generation fluoroquinolone.7 CIP has been continuously detected in water and wastewater at various concentration levels of 3–87 μg L−1 in the effluents from hospitals8–10 and at 31 mg L−1 in the wastewater from drug production facilities.8,11 In this study, CIP was selected as a model of a recalcitrant contaminant that forms a serious threat to the ecosystem and human health by proliferating bacterial drug resistance, even when found in small concentrations or existing as a low residual activity pollutant in the water system.12 Removal of CIP is complicated by its poor biodegradability and multifaceted structures; however, it can be removed through a photocatalytic method13–16 particularly when combined with advanced oxidation processes (AOPs), such as photocatalytic ozonation. Due to the synergetic effects of ozonation and photocatalysis, the efficiency of photocatalytic ozonation has proven to be higher than with either method alone.1,17 Ozone, as a strong oxidizer, can trap excited electrons from the conduction band (CB) of the photocatalyst, thereby restraining the recombination of photo-induced electrons and holes, which can further enhance the efficiency of photocatalysis. Furthermore, the ozonide ion radicals (O3˙−) generated in this process can be a good source of hydroxyl radicals.17 Therefore, photocatalytic ozonation can remove pollutants efficiently with high mineralization.17,18 Among various available semiconductors, titanium dioxide (TiO2), as a photocatalyst, has been the subject of much research.19 Some of the important advantages of TiO2 are chemical stability, high photoactivity, accessibility, and relative inexpensiveness. These properties make it a perfect candidate for the photocatalytic process.6,20 However, there are some drawbacks to using TiO2, including low adsorption, difficult recovery, fast electron–hole recombination, and the agglomeration phenomenon of suspended powder TiO2 at high loading, all of which limit TiO2 practical application in wastewater treatment.21,22 As a strategy to tackle these problems, immobilization of TiO2 nanoparticles on support materials can resolve several important issues. First, the problems related to filtering and recovering the photocatalyst can be resolved; second, the reduction of ultraviolet (UV) light intensity due to dispersion and absorption of the radiation by the catalyst particles can be minimized. The appropriate support should incorporate the following properties: chemical inertness, high specific surface area, and transparency to UV radiation.23,24 Recently, mesoporous supports with high surface areas, including silica, alumina, glass plate, zeolite, and clay have been considered for improving TiO2 photocatalytic activity.21,25–28 The related literature shows that montmorillonite (MMT) can be very useful in wastewater decontamination.29–31 Dispersion of TiO2 particles into layered MMT structures can stabilize TiO2 particles and take advantage of the surface of the TiO2 crystals accessible to various molecules.32 This availability leads to the improvement of the catalysts' performance. However, the principal shortcoming of AOPs is their high electricity requirements, which make their operating costs much higher than those of conventional treatment methods. More economical technologies that can be run by black-light or UVA light sources, or solar radiation have recently attracted researchers' attention.3,33,34 One efficient and economical photocatalytic ozonation system was the simultaneous utilization of UVA radiation with ozone in the presence of immobilized semiconductor photocatalysts, such as TiO2 nanoparticles.3 In the present study, CIP was used as the model antibiotic pollutant compound to evaluate the photocatalytic ozonation performance of a TiO2/MMT nanocomposite undergoing UVA radiation. The catalyst samples were synthesized through a hydrothermal method and characterized using SEM, EDX, HRTEM, FTIR, XRF, N2 adsorption–desorption, UV-vis DRS, and the pH of the zero point of charge (pHzpc) analyses. The mechanism of the photocatalytic ozonation process in the presence of various radical scavengers was thoroughly investigated. Finally, the degradation by-products generated in the photocatalytic ozonation process were analyzed by gas chromatography coupled to mass spectrometry (GC-MS). To the best of our knowledge, the photocatalytic ozonation of CIP, under the above-mentioned conditions, has not been previously investigated.
2. Experimental
2.1. Chemicals
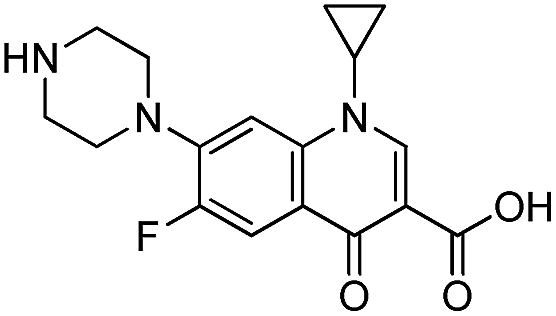
Montmorillonite K10 (MMT) was provided by Sigma-Aldrich Co. (USA). The cation exchange capacity (CEC) of the clay was 120 meq./100 g, as estimated by the ammonium acetate method.35 Cetyltrimethylammonium bromide (CTAB) was obtained from Sigma-Aldrich Co. (USA) too. Tetraethyl orthotitanate (TEOT), a precursor of Ti and N,O-bis-(trimethylsilyl) acetamide were purchased from Sigma-Aldrich Co. (USA) was purchased from Sigma-Aldrich Co. (USA). All other chemicals and reagents, which were of the analytical grade, were provided by Merck (Germany) and used without any further purification. All aqueous solutions were prepared using distilled water, which had been freshly prepared in the laboratory. Table 1 shows the structure and properties of CIP which was used as a model pollutant.
Table 1 Characteristics of the ciprofloxacin (CIP)
| Chemical structure |
Molecular formula |
Mw (g mol−1) |
λmax (nm) |
Therapeutic group |
Solubility in water (mg mL−1) |
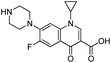 |
C17H18FN3O3 |
331.346 |
276 |
Antibiotic |
30 |
2.2. Synthesis of TiO2 and TiO2/MMT nanocomposite
A simple hydrothermal method was used to prepare the samples. The TiO2/MMT nanocomposite was synthesized as explained here. In a typical experiment, 1 g of MMT was swelled in 100 mL of distilled water and the mixture was stirred for 24 h at room temperature to obtain the uniform suspension. Then, 10 mmol of TEOT was added dropwise into 80 mL distilled water and stirred for 10 min. Subsequently, the desired amount of CTAB was added to the obtained solution in the dropwise manner under continuous stirring. The pH of the solution was adjusted to 11–12 with NaOH (0.1 M), and then it was stirred for 1 h. Then, the freshly prepared Ti solution was added to the MMT suspension. The mixture was sealed and stirred for another 1 h. After that, the well-mixed suspension was transferred into a Teflon-lined stainless-steel autoclave. The autoclave was sealed and maintained at 160 °C for 24 h, and then it was cooled at room temperature naturally. The resulting product was washed three times with distilled water and ethanol to remove any remaining residual impurities. The product was dried in an oven at 80 °C for about 24 h. The final powders were obtained accordingly. Pure TiO2 nanoparticles were synthesized by the same procedure without the addition of MMT.
2.3. Characterization
The chemical composition of the MMT and TiO2/MMT was determined using X-ray fluorescence (XRF) (Rigaku ZSX Primus II, Japan). Scanning electron microscope (SEM) equipped with an energy-dispersive X-ray spectroscopy (EDX) microanalysis was used to analyze the surface morphology and the elemental analysis of the samples was carried out using a Mira 3 FEG-SEM (Tescan, Czech Republic). Fourier transform infrared (FTIR) spectra were recorded on a Tensor 27, Bruker (Germany), in a wavenumber range of 4000–400 cm−1 and at a resolution of 1 cm−1, by using the KBr pellet technique. The high resolution transmission electron microscopy (HRTEM) images were taken using a JEOL JEM-2100F (Japan) operated at 200 kV. The ultraviolet-visible diffuse reflectance spectrometry (UV-vis DRS) of the samples were recorded using a Shimadzu UV-2550 (Japan) spectrophotometer. The microstructural properties of the samples were determined using N2 adsorption–desorption isotherms at 77 K on a Gemini 2385 nitrogen adsorption apparatus (Micromeritics Instruments, USA); using Brunauer–Emmett–Teller (BET) and Barrett–Joyner–Halenda (BJH) methods. UV-vis diffuse-reflectance spectra (DRS) of samples were recorded to determine the optical band gap (Eg) of pure TiO2 and TiO2/MMT samples. The band gap energy can be estimated from the intersection of the tangent along the absorption edge and wavelength axis. The reaction intermediates produced during the photocatalytic ozonation process were identified using a gas chromatograph (6890, Agilent Technologies, CA); this was done through a 30 m to 0.25 mm HP-5MS capillary column coupled with a mass spectrometer (5973, Agilent Technologies, Canada). To this aim, 500 mL of CIP solution with the concentration of 20 mg L−1 was treated for 15 min under the optimized conditions. The organic components of the treated solution were extracted with 40 mL of diethyl ether three times. The collected organic solution was evaporated and the remaining solid was dissolved in 100 μL of N,O-bis-(trimethylsilyl)-acetamide by heating at 60 °C and stirring for 10 min. The silylated products obtained in this way were then analyzed three times by GC-MS according to the following temperature settings: 50 °C for 4 min, 8 °C min−1 up to 300 °C, and the holding time of 4 min. The temperature of the inlet, transfer line and detector was 250, 250 and 300 °C, respectively.3
2.4. Experimental set-up and procedure
Individual oxidation and integrated photocatalytic ozonation experiments were conducted in a 1 L capacity cylindrical borosilicate glass reactor with a quartz cap in the batch mode. Fig. 1 shows the schematic of the reactor used in this study. The reactor was equipped with a magnetic stirrer, an inlet for ozone, a diffuser, a sampling point and an outlet for the non-absorbed ozone gas. The reactor walls were first covered with aluminum foil and then an insulating material to prevent the release of radiation and heat to the surroundings. Ozone was made from the pure oxygen using an ozone generator (Triogen, Scotland). We used one triple valve to control the flow of ozone. An oxygen–ozone mixture was continuously bubbled into the solution through a diffuser located at the bottom of the reactor. The gas flow rate was changed from 1 to 6 L h−1 with ozone dissolved concentration being between 0.04 and 0.33 mg L−1 (Table S1†). 2 L h−1 as optimum gas flow rate was used in all trials. Ozone inlet concentration was fixed at about 5 mg L−1 in the ozone generator. Dissolved ozone concentration was determined by the indigo method using a UV-vis spectrophotometer.36 Artificial irradiation was provided by an 8 W UVA lamp (Philips, the Netherlands with wavenumber region of 315–400 nm) placed inside a quartz tube within the reactor. Before the reaction under UV or ozone, the suspension was magnetically stirred in darkness for 10 min to establish an adsorption–desorption equilibrium. In each experiment, 500 mL of the reaction mixture containing definite concentrations of CIP and catalyst (TiO2 and TiO2/MMT) was prepared. The initial pH was set to the desired value with HCl and NaOH (0.1 M); also, no buffer solution was used to avoid any complexity (changes of solution pH during reaction were not significant). The solution pH was regulated using a Mettler Toledo pH meter (China). At time intervals of 3 min, the samples were withdrawn from the reactor and sodium sulfite was added to them to stop any development of ozone or hydroxyl radical oxidation. The suspended photocatalyst particles were removed by centrifuging for 10 min at 6000 rpm (Hettich EBA 20, Germany). The relative concentration of the CIP solution was calculated by the relative absorbance. In addition, the residual concentration of CIP was determined using Varian Cary 100 UV-vis spectrophotometer (Australia) at the maximum wavelength of 276 nm. Degradation efficiency (%) = [(A0 − A)/A0] × 100, was used to determine the percent of degradation of CIP, where A0 and A are the absorbance at the beginning and the definite time of the process, respectively. For measuring the pH zero point of charge (pHzpc) of TiO2/MMT nanocomposite 500 mL 0.01 M NaCl was prepared and divided into eight solutions with the pH range of 3 to 10. Then, 0.2 g of the catalyst was added to each solution. Finally, the final pH of each solution was measured after 48 h shaking and plotted against the initial pH to determine the pHzpc of the catalyst.37 The intersection point between pHfinal and pHinitial was recorded as pHzpc. Various organic and inorganic ions, acting as scavengers, were added to an aqueous solution in the photocatalytic ozonation process to investigate the mechanism of the process. A 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio of scavengers to CIP was tested. The reusability of TiO2/MMT nanocomposite was evaluated by repeating the batch test. Prior to each reuse, the entire liquid phase of the previous use was removed and the new solution was added without any catalyst treatment.
:1 molar ratio of scavengers to CIP was tested. The reusability of TiO2/MMT nanocomposite was evaluated by repeating the batch test. Prior to each reuse, the entire liquid phase of the previous use was removed and the new solution was added without any catalyst treatment.
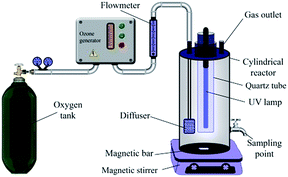 |
| | Fig. 1 Schematic representation of the reactor used for different oxidative processes. | |
3. Results and discussion
3.1. Catalyst characterization
The chemical composition of MMT and TiO2/MMT were determined by using XRF analysis. The data presented in Table S2† shows that the main structure and textural properties of the MMT sample were SiO2 and Al2O3, while the content of other oxide components (Fe2O3, K2O, MgO, CaO, Na2O and TiO2) were less than 5 wt%. According to the obtained results, the TiO2 content was increased to 58.50 wt% in the TiO2/MMT nanocomposite. The content of SiO2 and Al2O3 was decreased considerably after the immobilization of TiO2 nanoparticles on the surface of, or in between, the MMT layers. These results confirmed that a considerable amount of TiO2 species entered into the layered structure of the MMT and hybridized with it, through a hydrothermal process. SEM analysis evaluated the surface morphology of the MMT, TiO2, and TiO2/MMT samples. The results are shown in Fig. 2. As depicted, MMT had an uneven structure with a non-uniform size distribution. In the SEM image of the MMT sample, some phase separation and cracks were observed, which seemed to be a heterogeneous surface morphological element (Fig. 2a). As shown in Fig. 2b, TiO2 nanoparticles were irregular in size and shape. This non-uniformity can be related to the aggregation of synthesized TiO2 nanoparticles and the growth of irregular crystalline grains during synthesis. Fig. 2c shows the SEM image of a TiO2/MMT nanocomposite. As displayed, there is an obvious change in the MMT morphology, indicating that the MMT layers are destroyed and good dispersion of TiO2 nanoparticles on the MMT plates is occurred. The size distribution of TiO2 particles in the TiO2/MMT sample was determined using manual microstructure distance measurement software (Nahamin Pardazan Asia Co., Iran). Fig. 2d exhibits the distribution of TiO2 particles sizes within the TiO2/MMT sample, which indicates that most of the TiO2 nanoparticles are in the range of 40–50 nm with a frequency of 29%. The immobilization of TiO2 nanoparticles on the surface of or between MMT layers reduced the aggregation of TiO2 nanoparticles and increased the shape uniformity of the synthesized nanoparticles.
 |
| | Fig. 2 SEM micrographs of (a) MMT, (b) TiO2, (c) TiO2/MMT, and (d) distribution of TiO2 particles size in TiO2/MMT sample. | |
EDX analysis was carried out to determine the composition of the TiO2/MMT nanocomposite. The EDX analysis results for the TiO2/MMT sample are represented as numerical values in Table S3.† Based on the EDX results, the TiO2/MMT contained C, O, Si, Ti, Mg, Al, Na, and K, which clearly shows that Ti species are formed in the interlayer and on the surface of the MMT support. The ratio of Si:Al is 2.38 (∼2.0), which can be a typical value for silica–alumina sheets in a 2:1 ratio within the structure.38 The elemental gold in the EDX results can be due to the gold plating on the sample that prevents static discharging.
FTIR analysis is an effective technique used to identify surface-active functional groups within materials. A comparison of the FTIR spectra of MMT, TiO2, and TiO2/MMT samples were attempted, and the results are displayed in Table 2.6,21,29,39 In the spectrum of TiO2 nanoparticles, the typical characteristic bands at 729 cm−1 and 800 cm−1 contributed to the vibration absorption peak of O–Ti–O and Ti–O, respectively.40 The bands at 1630 cm−1 can be primarily attributed to the deformation vibration of H–O–H bonds of the physisorbed water.6,41 The peak at 3420 cm−1 can be assigned to the surface functional groups –OH of nanoparticles.6 The band at the interval of 2800–3000 cm−1 can be related to –CH– stretching vibration that appears in TiO2 nanoparticles due to the introduction of the CTAB surfactant. Compared with MMT, the new absorbance peak at 935 cm−1 of the Ti–O–Si revealed that TiO2 nanoparticles were immobilized onto the MMT clay. Meanwhile, the absorption bands of the –OH stretching vibrations of the physically adsorbed water were shifted, possibly as a result of the interaction between the MMT and TiO2.
Table 2 Some fundamental FTIR absorption frequencies of MMT, TiO2, and TiO2/MMT
| MMT |
TiO2 |
TiO2/MMT |
| Position (cm−1) |
Assignments |
Position (cm−1) |
Assignments |
Position (cm−1) |
Assignments |
| 472 |
Si–O–Si deformation |
480 |
Ti–O stretching |
462 |
Si–O–Si bending vibrations |
| 530 |
Al–O–Si deformation |
729 |
O–Ti–O stretching |
518 |
Si–O asymmetric stretching |
| 798 |
Al–O stretching |
800 |
Ti–O stretching |
935 |
Ti–O–Si stretching |
| 1050 |
Si–O asymmetric stretching |
1466 |
Ti–O–Ti stretching |
1047 |
Si–O stretching |
| 1630 |
H–O–H interlayer |
1630 |
H–O–H interlayer |
1471 |
Ti–O–Ti stretching |
| 3420 |
–OH stretching of water |
2845 |
C–H symmetric stretching |
1638 |
Ti–OH stretching |
| 3626 |
–OH stretching of crystalline water |
2924 |
C–H asymmetric stretching |
2850 |
C–H symmetric stretching |
| |
|
3420 |
–OH stretching |
2921 |
C–H asymmetric stretching |
| |
|
|
|
3420 |
–OH stretching |
| |
|
|
|
3622 |
–OH stretching of crystalline water |
The morphology of the MMT and TiO2 nanoparticles synthesized on the MMT were also investigated through HRTEM. The HRTEM images of samples are shown in Fig. S1.† The remarkable difference between the HRTEM images of the pure MMT and TiO2/MMT nanocomposite can be clearly observed. As depicted in Fig. S1a and b,† the MMT clay support has a very smooth, undulating, and multilayered structure. Fig. S1c† shows that the synthesized TiO2 nanoparticles are well-supported on the MMT surface. The black dots on the surface of the MMT support are the TiO2 particles. The HRTEM image of TiO2/MMT nanocomposite clearly shows a disorder in the stacking layers of the MMT support, which can be ascribed to the presence of TiO2 nanoparticles between them. In other words, an interaction between the support MMT and the TiO2 nanoparticles can be inferred from the HRTEM images of the TiO2/MMT nanocomposite, which is predictable according to uniform TiO2 nanoparticles dispersion (Fig. S1c†). To obtain further information regarding the specific surface area, pore size and pore volume, N2 adsorption–desorption experiments were carried out and the results were analyzed using the Brunauer–Emmett–Teller (BET) and Barrett–Joyner–Halenda (BJH) adsorption isotherms. Fig. S2† displays the N2 adsorption–desorption isotherms and their corresponding pore size distribution curves (inset) for the MMT, TiO2 and TiO2/MMT samples. As can be seen from Fig. S2a,† the adsorption isotherm of MMT is type II with a hysteresis loop, characterizing a mesoporous texture with a uniform pore size distribution. At high relative pressure from 0.45 to 0.98, the isotherms exhibited the hysteresis loops of an H4 type, revealing that the sample contained open slit-shaped capillaries.42 This type of isotherm can be related to multilayer physical adsorption, describing the strong interaction between the adsorbate and the adsorbent. As depicted in Fig. S2a,† the adsorption branch of isotherms at high relative pressure (more than 0.45) is inconsistent with the desorption branch due to the capillary condensation resulting in a hysteresis loop.43 Meanwhile, the hysteresis loop was closed at a low relative pressure for MMT and TiO2 samples, but it was opened for the TiO2/MMT nanocomposite. The lack of total closure of the hysteresis loop at low relative pressure was the result of the swelling effect.43 Nevertheless, the N2 adsorption–desorption isotherms for both TiO2 and TiO2/MMT samples were quite different from those for the MMT support (Fig. S2b and c†). The isotherms for the TiO2 and TiO2/MMT were type IV with a pronounced H3-type hysteresis loop, showing relative pressure in the range of 0.45 to 1. This signifies that the samples have a lamellar pore structure, a disordered slit, and a wedge shape.44 The position of the inflection point of P/P0 can be related to a diameter in the mesoporous range, and the sharpness of the step shows the uniformity of the mesopore size distribution.45 As observed in Fig. S2c,† the sharpness of the steps of the inflection curves of TiO2/MMT were lower than those of MMT, thereby indicating the incorporation of TiO2 nanoparticles into the MMT support interlayer galleries.46 For MMT isotherms, adsorption and desorption lines overlapped in the low relative pressure range, while the hysteresis loop existed in the high relative pressure region. From a comparison of all isotherms demonstrated in Fig. S2,† a large amount of adsorption and a wider hysteresis loop were found for MMT and TiO2/MMT, respectively. The pore size distribution of the samples, as estimated according to the BJH method from the adsorption branch, is shown in Fig. S2† (inset). The average pore size radius of the MMT, TiO2 and TiO2/MMT samples was 3.175, 6.765 and 4.993 nm, respectively. Additionally, the BET surface areas for the MMT, TiO2 and TiO2/MMT were 279.278, 8.813 and 53.058 m2 g−1, with the corresponding BJH adsorption pore volumes of 0.456, 0.035 and 0.105 cm3 g−1, respectively. There was a considerable decrease in the surface area and pore volume, as compared to the MMT support, which was due to the blocking of the pores by the TiO2 nanoparticles and the collapsing of the mesostructure. Comparatively, the BET surface area of the TiO2/MMT nanocomposite was much higher than that of the TiO2 nanoparticles, thereby suggesting the TiO2 nanoparticles were widely dispersed over the MMT support.
To evaluate the effects of the immobilization of TiO2 nanoparticles onto the MMT support on the optical properties of TiO2/MMT nanocomposite, UV-vis DRS analysis was launched. Fig. 3 shows the corresponding UV-vis DRS spectra for the MMT, TiO2 and TiO2/MMT samples. The absorption thresholds for the samples were obtained from the UV-vis DRS curves by extrapolating the tangent lines of the spectra. Bare TiO2 nanoparticles show absorption in the UV region without any absorption in the visible range. As depicted in Fig. 3a, the threshold wavelength for the synthesized TiO2 nanoparticles is about 387 nm. After the immobilization of TiO2 onto the MMT, the absorption threshold is shifted to 375 nm. Compared to the pure TiO2, a blue shift of the absorption edges towards the ultraviolet region was observed in the TiO2/MMT nanocomposite. This feature resulted from the quantum-size effect, that is, the TiO2 nanoparticles deposited on the MMT support are small enough to show the quantum-size effect.47,48 Accordingly, the TiO2/MMT nanocomposite possess showed much improved photoactivity capability under UV light irradiation. The light absorption of the pure MMT was increased, such that it became almost transparent in wavelengths longer than 300 nm, which was consistent with the results reported in the literature.47–49 The MMT showed weak absorption of UV light. The band gap energies (Eg) of the samples were calculated using the equation (Ahν)2 = K(hν − Eg), where hν is the energy of a photon (eV); A is the absorption coefficient; K is a constant, and Eg is the band gap. The band gap can be calculated by extrapolating the linear part of the spectra in a diagram of (Ahν)2 versus the photon energy (Fig. 3b). The band gap energy values for the TiO2 and the TiO2/MMT nanocomposite were calculated as 3.20 and 3.30 eV, respectively, implying that immobilization of TiO2 nanoparticles onto the MMT support increased the optical band gap energy; therefore, the electron–hole separation occurred relatively better within the TiO2/MMT nanocomposite.
 |
| | Fig. 3 (a) UV-vis DRS absorbance spectra and (b) plot of (αhν)2 versus hν for the samples. | |
3.2. Comparison of different processes in removing CIP
Before beginning the functional evaluation of the parameters, a series of experiments were carried out to compare the removal efficiency of the CIP through different processes. Fig. 4 shows the effect of CIP degradation efficiency on the reaction time for each process. For example, CIP degradation is low (10%) when using direct photolysis under UVA radiation in the presence of oxygen. A controlled adsorption experiment was then carried out. The removal of CIP through adsorption onto TiO2 was found to be 15% in 30 min, indicating that adsorption had no significant contribution in the removal of CIP. As also shown in Fig. 4, the degradation of CIP in 30 min is 22.08%, using a photocatalytic process. The photocatalytic oxidation process acts rather slowly and required more reaction time to complete the degradation of the pollutant. Further, the degradation efficiency of CIP by ozonation and photolytic ozonation (UV/O3) at 30 min is 67.92% and 71.76%, respectively. CIP degradation by ozonation and photolytic ozonation takes place exclusively in the liquid phase through both direct and indirect mechanisms. Based on the literature review, the organic compound at the natural pH of the solution can be removed through both the direct and indirect mechanisms, where hydroxyl radicals are generated by ozone decomposition. However, the indirect mechanism is favored in alkaline media. Therefore, CIP degradation is implemented through the ozonation process as shown in eqn (1)–(10).3,4| | |
O3 + CIP → degradation products + O3˙− (direct)
| (1) |
| | |
O3 + OH− → HO2− + O2 (indirect)
| (2) |
| | |
O3 + HO2− → HO2˙ + O3˙− (indirect)
| (3) |
| |
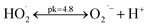 | (4) |
| | |
O2˙− + O3 → O3˙− + O2
| (7) |
| | |
HO3˙ + HO3˙ → 2O2 + H2O2
| (9) |
| | |
˙OH + HO3˙ → O2 + H2O2
| (10) |
 |
| | Fig. 4 Comparison of the degradation efficiency of CIP with different processes. (a) Photolysis; (b) adsorption; (c) photocatalytic oxidation; (d) ozonation; (e) photolytic ozonation; (f) catalytic ozonation; and (g) photocatalytic ozonation. Conditions: [CIP]0 = 20 mg L−1, [catalyst]0 = 0.04 g L−1, ozone gas flow rate = 2 L h−1 and pH = 5. | |
In photolytic ozonation, ozone photolysis quantum yield and molar absorptivity are not significant in the UVA region.50 However, a slight increase in the degradation efficiency of CIP by the photolytic ozonation process can be related to the photolysis of ozone molecules by UVA radiation, because ozone absorbs some of the incoming photons; this absorption results in the decomposition of ozone into atomic oxygen and H2O2, as shown in eqn (11) and (12).3,50
| | |
O3 + hν → O(1D) + O2
| (11) |
According to eqn (12), atomic oxygen reacts with water, producing hydrogen peroxide, forming a new reactive species through the subsequent reactions eqn (13) and (14), and leading to the enhanced degradation of target pollutants.51
| | |
O(1D) + H2O → ˙OH + ˙OH
| (14) |
As observed in Fig. 4, the degradation efficiency of CIP using catalytic ozonation (80.14% at 30 min) is notably higher than that of ozonation. This result shows the implementation of the heterogeneous catalytic ozonation on the surface of the photocatalyst. More importantly, CIP degradation dramatically increased when coupling photocatalytic oxidation with ozonation. The final degradation efficiency of CIP using the photocatalytic ozonation process is 90.00% in 30 min, which is 67.92% and 22.08% higher than the results obtained using either photocatalytic oxidation or ozonation, respectively. This result indicates that the TiO2/MMT photocatalyst triggered a synergistic effect between photocatalysis and photolytic ozonation that enhanced the efficient degradation of CIP. All possible reaction steps during the photocatalytic ozonation of CIP are explained by eqn (15)–(22).
| | |
TiO2 + hν → eCB− + hVB+
| (15) |
| | |
MMT(metals) + eCB− → MMT(metals) − e−
| (17) |
| | |
hVB+ + CIP → products
| (21) |
| | |
˙OH + CIP → products
| (22) |
In addition, the photo-generated electrons can be trapped with dissolved oxygen, as shown by eqn (23), resulting in the production of superoxide anion O2˙− and the formation of hydroxyl radicals as seen in eqn (24)–(28); this process, in turn, can improve the degradation of the pollutant:3
| | |
O2˙− + O3 → O3˙− + O2
| (24) |
| | |
H2O2 + O2˙− → ˙OH + OH− + O2
| (27) |
| | |
H2O2 + eCB− → ˙OH + OH−
| (28) |
In addition, as can be seen in eqn (29)–(31), metals ions in MMT may react with the superoxide species and suppress the recombination of electrons–holes, thereby increasing the degradation efficiency of CIP.52
| | |
MMT(metals) + O2− → MMT(metals) − e− + O2
| (29) |
| | |
O2˙− + CIP → degradation of CIP
| (31) |
Eqn (32) quantifies the synergistic factor within the photocatalytic ozonation process.
| |
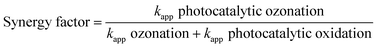 | (32) |
Also, the apparent pseudo-first-order reaction rate constant (kapp) for each process can be calculated from the slope of the plot of ln(A0/A) against process time (t) (Table 3). The synergy factor was found to be 1.6. This factor can be explained by referring to the established mechanism investigated in studies focusing on the degradation of different organic pollutants.3,53,54 The considerable enhancement in the degradation rate of organic pollutants through employing photocatalysis in combination with ozone is due to the generation of more ˙OH radicals on the TiO2 surface through the formation of ozonide anion radicals O3˙− (eqn (15) and (18)–(20)). Because the trapping of photogenerated electrons using ozone is more efficient, the recombination between holes and electrons is minimized. Consequently, a larger number of radicals are produced, leading to the acceleration of the reaction. While molecular oxygen accepts the photogenerated electrons, the resulting superoxide radical anion can react with ozone, giving hydroxyl radicals in the later steps (eqn (19)–(24)). As a result, when the photocatalyst is irradiated in the presence of ozone, a greater number of ˙OH radicals are produced, in comparison with the same process in the presence of oxygen.
Table 3 Degradation efficiency (DE%), (except for adsorption) apparent pseudo-first-order constants and correlation coefficients of the utilized processes for CIP degradation
| Processes |
DE (%) |
kapp (min−1) |
R2 |
| Photolysis |
10.00 |
0.0031 |
0.9432 |
| Adsorption |
15.01 |
0.0044 |
0.8341 |
| Photocatalytic oxidation |
22.08 |
0.0072 |
0.9015 |
| Ozonation |
67.92 |
0.0362 |
0.9941 |
| Photolytic ozonation |
71.76 |
0.0398 |
0.9909 |
| Catalytic ozonation |
80.14 |
0.0499 |
0.9909 |
| Photocatalytic ozonation |
90.00 |
0.0688 |
0.9846 |
3.3. The effect of TiO2/MMT dosage
Catalyst dosage is an important parameter in the photocatalytic ozonation process. To evaluate the effect of the catalyst dosage on CIP degradation during photocatalytic ozonation, the TiO2/MMT dose can be varied from 0.005 g L−1 to 0.08 g L−1. As depicted in Fig. 5, the degradation efficiency of CIP in photocatalytic ozonation gradually increased with an increase in the catalyst dosage up to 0.006 g L−1 and then slightly decreased. However, when the catalyst dosage is in the range of 0.005–0.04 g L−1, the degradation efficiency of CIP increased more quickly, when compared to that of the dosing in the range of 0.04–0.06 g L−1. The increase of the catalyst dosage demonstrates two opposing factors contributing to the degradation process. First, the number of active sites adsorbed on the surface of the particles increased with the increase of the catalyst amount, thereby resulting in the enhanced degradation efficiency. This, in turn, produced more hydroxyl and superoxide radicals.55 On the other hand, a scattering phenomenon emerges, leading to the generation of e−–h+ pairs on the surface of the catalyst, lost efficacy, and decreased degradation efficiency.56 The degradation efficiency of CIP at 0.04 g L−1 of TiO2/MMT is 19.25% higher than at 0.005 g L−1. Meanwhile, the degradation efficiency of CIP is found to be slightly increased when the dose of TiO2/MMT increased from 0.04 g L−1 to 0.06 g L−1. Under comprehensive consideration, a catalyst dosage of 0.04 g L−1 was selected as the best dosage of the catalyst in the subsequent experiments.
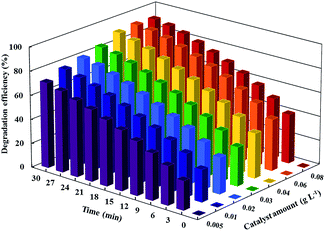 |
| | Fig. 5 The effect of catalyst amount on the degradation efficiency of CIP. Conditions: [CIP]0 = 20 mg L−1, ozone gas flow rate = 2 L h−1 and pH = 5. | |
3.4. The effect of the initial CIP concentration
The effect of CIP concentrations on CIP degradation during the photocatalytic ozonation process was investigated by changing CIP concentrations in the range of 5–25 mg L−1. The experimental results are presented in Fig. 6. In addition, the kinetic data of the experiments, using various CIP concentrations, are listed in Table S3.† According to the results presented in Table S3,† the degradation rate constant for CIP is increased from 0.115 mg L−1 min−1 to 0.6587 mg L−1 min−1 when the initial concentration of CIP is enhanced from 5 to 25 mg L−1. The increase in the oxidation rates can be attributed to the quantitative increase in the reaction amongst the generated hydroxyl radicals and CIP molecules at the higher concentration of CIP. A similar behavior is reported by other researchers as the increase in the initial pollutant concentration resulted in an increase in the oxidation rates and the degradation of various organic pollutants in the aqueous phase.57,58
 |
| | Fig. 6 The effect of the initial CIP concentration of the degradation efficiency by photocatalytic ozonation. Conditions: [catalyst]0 = 0.04 g L−1, ozone gas flow rate = 2 L h−1 and pH = 5. | |
3.5. The effect of ozone gas concentration and flow rate
According to the related literature,3,51 increasing the ozone concentration in the inlet gas can lead to more effective degradation of organic pollutants in the photocatalytic ozonation process. The effect of ozone concentration within the inlet gas on the degradation efficiency of CIP was investigated by changing the ozone gas flow rate from 1 L h−1 to 6 L h−1. The dissolved concentration of ozone is shown through each applied ozone gas flow rate in Table S1.† The results are presented in Fig. 7. As shown, the photocatalytic ozonation efficiency is significantly enhanced when the ozone gas flow rate is increased from 1 L h−1 to 2 L h−1, while further improvement of the gas flow rate makes no significant difference. This factor indicated that there was not enough ozone for the scavenging photogenerated electrons when the ozone gas flow rate fell below 2 L h−1. Fig. 7 reveals that the ozone gas flow rate of 2 L h−1 greatly expedites the degradation of CIP during the photocatalytic ozonation process. This phenomenon can be caused by a synergistic effect, as it leads to an increase in the production of more reactive oxygen species (ROS), particularly hydroxyl radicals.59 Indeed, by increasing the ozone gas flow rate, the concentration of the dissolved ozone is increased. Additionally, the increase of the ozone gas flow rate led to an increase in the agitation of the reaction medium. The dissolved ozone molecules can be easily adsorbed on the surface of the photocatalyst in its molecular or dissociative form through either physical adsorption or the formation of weak hydrogen bonds with the surface hydroxyl group. Ozone molecules capture electrons produced on the photocatalyst surface yielding ozonide radical anion; consequently, the produced ozonide radical anion (O3˙−) results in a high number of hydroxyl radicals' formation and the increase in the degradation efficiency of CIP.60,61 Our research findings in the present study were consistent with those of previous studies in the related literature.57,62,63
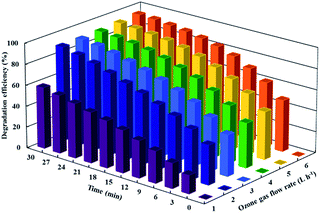 |
| | Fig. 7 The effect of the inlet ozone flow rate on the photocatalytic ozonation of CIP. Conditions: [CIP]0 = 20 mg L−1, [catalyst]0 = 0.04 g L−1 and pH = 5. | |
3.6. The effect of the initial pH
pH enacts the degradation efficiency of organic pollutants.64 Therefore, this parameter was chosen as one of the main process variables investigated in the present study. The effect of the initial pH on the degradation efficiency of CIP in the photocatalytic ozonation process is studied in the pH 3–11 range (Fig. 8). As observed, the degradation efficiency of CIP is enhanced by increasing the solution pH to 5 and then decreased. The effect of pH on the photocatalytic ozonation process is complex; it performs multiple roles in the electrostatic interaction between the photocatalyst surface, solvent molecules, and the substrate during the reaction process.6 CIP has two pKa values (5.9 and 8.89). According to the literature, CIP can exist as a cation (CIP0,+), a zwitterion (CIP−,+), or an anion (CIP−,0) at different pHs.8,65 The pH at which the surface of a catalyst is uncharged is known as the zero point charge (pHzpc). The pHzpc of the TiO2/MMT nanocomposite was evaluated to better understand the surface charge, and the pHzpc of TiO2/MMT was 8.4. This reveals that the surface of TiO2/MMT photocatalyst has a positive charge at pH < 8.4 and a negative charge at pH > 8.4. When pH is lower than 5.9, CIP molecules have a positive charge, while CIP retains a negative charge when the pH value is higher than 8.9. Therefore, at low pH values (around 3), the decrease in the degradation efficiency of CIP may be attributed to the repulsing effect between the positively charged surfaces both of the catalyst and CIP. On the other hand, the surface of TiO2/MMT and CIP molecules are negatively charged under basic conditions, thereby resulting in charge repulsion and the lowest degree of adsorption and degradation efficiency. At pH 5 (the natural pH of CIP), the surface of photocatalyst remains positive with the CIP is in its neutral form; under these conditions, increased adsorption leads to the enhanced degradation efficiency of CIP. In other words, when the pH is increased from 3 to 5, proportions of CIP neutral form are increased, thereby increasing the interaction with the positively charged photocatalyst. Accordingly, the synergistic effects of ozone on the TiO2/MMT photocatalyst were observed at CIP original pH 5.3,18 Considering the results, the experiments were carried out at pH 5.
 |
| | Fig. 8 The effect of the initial pH value on the photocatalytic ozonation of CIP. Conditions: [CIP]0 = 20 mg L−1, [catalyst]0 = 0.04 g L−1 and ozone gas flow rate = 2 L h−1. | |
3.7. The effect of various scavengers on CIP degradation
To investigate the degradation mechanism, including the effects of surface reactions and the main ROS involved in CIP degradation within the photocatalytic ozonation system, a series of experiments were performed using various inorganic ions and ROS inhibitors, such as NaF, NaH2PO4, Na2SO4, Na2CO3, NaNO3, NaCl, and KI. The concentration each of inorganic scavenger added to the solution was kept constant at 20 mg L−1. The obtained degradation rate constants and the degradation efficiency of CIP, with or without the addition of various inorganic ions, along with their corresponding regression coefficients, are shown in Table 4. In comparison with the control test of the aqueous CIP solutions without the presence of the scavenger, the existence of all anions reduces the CIP degradation rate constant.
Table 4 The effects of some ROS inorganic scavengers on the degradation rate constant of 20 mg L−1 CIP after 30 min of photocatalytic ozonation at the ozone flow rate of 2 L h−1 and the pH of 5
| Scavenger |
DE (%) |
kapp (min−1) |
R2 |
| No scavenger |
90.00 |
0.0688 |
0.9846 |
| NaF |
82.55 |
0.0540 |
0.9908 |
| Na2CO3 |
80.85 |
0.0534 |
0.9964 |
| NaH2PO4 |
77.06 |
0.0466 |
0.9897 |
| Na2SO4 |
73.20 |
0.0459 |
0.9954 |
| NaCl |
74.45 |
0.0449 |
0.9941 |
| NaNO3 |
71.68 |
0.0441 |
0.9964 |
| KI |
35.19 |
0.0141 |
0.9954 |
Table 4 shows that the CIP degradation rate constant decreases slightly with the addition of NaF. This decrease of the degradation rate constant in the presence of F− ions can be attributed to the stronger adsorption ability of the F− ions on the surface of the photocatalyst.3 Therefore, the presence of F− ions indicated that the domination of these ions prevents the development of synergistic reactions, subsequently decreasing the production of hydroxyl radicals. Overall, these results indicated that the adsorption of the CIP and ozone molecules on the photocatalyst surface can be an important step in the photocatalytic ozonation process.3,66
In addition, the effect of NaH2PO4, which is a well-known inhibitor of ˙OH radicals1,3 on CIP degradation rate constant was also investigated. The results indicated that the presence of dihydrogen phosphate anions significantly reduces CIP degradation rate constant. The deleterious effects of H2PO4− can be described by the competitive adsorption of H2PO4− ions within the substrate onto the surface of the photocatalyst. These anions react with the hydroxyl radicals according to eqn (33),67 leading to inorganic radicals with lower reactivity, so the produced radicals do not participate in the degradation of CIP.
| | |
H2PO4− + ˙OH → HPO4˙− + H2O
| (33) |
Table 4 shows the degradation rate constant and the degradation efficiency of CIP during photocatalytic ozonation in the presence of Na2SO4. In the presence of sulfate ions in the aqueous solution, the degradation rate constant is decreased. The observed retardation effect of sulfate ions can be attributed to the adsorbed sulfate ions on the photocatalyst surface and to their reaction with photogenerated holes and hydroxyl radicals.6,68
| | |
SO42− + ˙OH → SO42− + OH−
| (35) |
The effect of CO32− ions on the photocatalytic ozonation activity of the catalysts is investigated by the addition of H2CO3 to the solutions of CIP. According to eqn (36), carbonate ions react with hydroxyl radicals, producing carbonate radicals69,70 as weak oxidizing agents, with a possible influence on the adsorption of the degrading species.
| | |
CO32− + ˙OH → CO3− + OH−
| (36) |
The effect of nitrate NO3− ions on the degradation rate constant of CIP is studied by adding NaNO3 into the CIP solution (Table 4). A detrimental effect is observed for the CIP degradation rate constant. The reaction of the adsorbed NO3− with positive holes (h+) and hydroxyl radicals inhibits the CIP degradation rate constant.71
| | |
NO3− + ˙OH → NO3˙ + OH−
| (38) |
Many studies have already reported that Cl− ions from NaCl added to the CIP solution can scavenge h+ and ˙OH via the following general reactions:6,52,71–73
| | |
Cl− + ˙OH → Cl˙ + H2O
| (40) |
| | |
ClOH˙ + H+ → Cl˙ + H2O
| (41) |
Such radicals can be induced to return to their chloride ions using electrons, thereby decreasing the availability of the holes, electrons, and ˙OH69 Alternatively, chlorine and dichloride anion radicals can further react with organic compounds through the addition/elimination reaction.69 As shown in Table 4, the presence of Cl− ions can significantly reduce the degradation rate constant of CIP.
KI has been used as a scavenger of hydroxyl radicals, holes and electrons.74–77 In the presence of I− ions, the degradation rate constant of CIP is significantly suppressed. When iodide ions are used as a diagnostic tool for suppressing the holes and the ˙OH process, the degradation rate constant of CIP remains largely inhibited, as represented in Table 4. The reaction of the adsorbed I− with holes and electrons has also been found to inhibit CIP degradation rate constant, as shown by eqn (43) and (44):
Therefore, the order of the inhibitory effect of the various inorganic scavengers can be described as follows: iodide > nitrate > chloride > sulfate > dihydrogen phosphate > carbonate > fluoride.
In addition to the inorganic scavengers, the effects of some organic scavengers on the degradation efficiency of CIP were evaluated. The photocatalytic ozonation of CIP was carried out in the presence of various organic scavengers, such as benzoquinone (BQ) (O2˙− scavenger), EDTA (h+ scavenger), CHCl3 (O2˙− scavenger), t-BuOH (˙OH scavenger), and L-ascorbic acid (O2˙− scavenger). The 1:1 molar ratio of scavengers to CIP was also tested. The obtained pseudo-first-order rate constant and degradation efficiency, with or without the addition of various organic scavengers along with their corresponding regression coefficients, are given in Table 5. The CIP degradation rate constant and the degradation efficiency are consistently reduced with the addition of scavengers into the reaction system. The observed decrease in the CIP degradation rate constant suggests the role of the corresponding active species in the degradation of CIP. The degradation rate constants for CIP are as follows: 0.0207 min−1, 0.0370 min−1, 0.0424 min−1, 0.0490 min−1 and 0.0550 min−1 for added CHCl3, with the addition of BQ, ascorbic acid, t-BuOH, EDTA and CHCl3, respectively. When BQ was added into the reaction system, the degradation rate constant of CIP was greatly restrained, as compared to the reaction without scavengers. A similar obvious suppression phenomenon was also observed in the case of the ascorbic acid. Therefore, O2˙− can be conclusively shown to cause the degradation of CIP through the photocatalytic ozonation process. However, the degradation rate constant of CIP in the presence of CHCl3, which is also an O2˙− scavenger, is slightly lower than that of the CIP reaction rate constant without scavengers, thereby indicating a weak scavenger effect of CHCl3 in comparison with BQ and L-ascorbic acid. Even with the lowered scavenger effect of CHCl3, this factor also demonstrates that O2˙− participates in the degradation of CIP during the photocatalytic ozonation process. Likewise, the degradation rate constant of CIP is decelerated in the presence of EDTA. The CIP degradation rate constant and degradation efficiency are both suppressed in the presence of EDTA, with the removal rate of approximately 79.86%, also confirming a partial role of holes in the degradation of CIP during the photocatalytic ozonation process. Moreover, due to its competitive adsorption, EDTA is likely to inhibit the degradation rate constant of CIP. Therefore, the significant inhibitory effect of EDTA can be attributed to both the competitive adsorption, which hinders the synergistic reactions, and the consumption of holes.3 While the degradation rate constant of CIP is significantly decreased, the reaction does not completely consume CIP in the presence of EDTA, probably due to the fact that direct ozonation is involved in the degradation of CIP during the photocatalytic ozonation process.3 Overall, our findings stand in good agreement with those of prior research.78,79
Table 5 The effects of some ROS organic scavengers on the degradation rate constant of 20 mg L−1 CIP after 30 min of photocatalytic ozonation at the ozone flow rate of 2 L h−1 and the pH of 5
| Scavenger |
DE (%) |
kapp (min−1) |
R2 |
| No scavenger |
90.00 |
0.0688 |
0.9846 |
| CHCl3 |
83.59 |
0.0550 |
0.9882 |
| EDTA |
79.86 |
0.0490 |
0.9909 |
| t-BuOH |
72.45 |
0.0424 |
0.9986 |
| L-Ascorbic acid (AsA) |
67.12 |
0.0370 |
0.9974 |
| BQ |
46.89 |
0.0207 |
0.9992 |
3.8. The possible mechanism for the photocatalytic ozonation process
Based on the data obtained, the degradation mechanism of CIP through the photocatalytic ozonation process is proposed in Fig. 9. The enhancement that occurs in the degradation efficiency of CIP through the immobilization of TiO2 nanoparticles on the surface of MMT can be due to the increased electron–hole separation time and the large surface area of the catalyst. The increase in the surface area leads to the more formation of active sites for photoreaction and adsorption, efficient light capturing; this, in turn, increases ROS production and consequently efficient degradation of CIP. In the photocatalytic ozonation process using TiO2/MMT catalyst, following the absorption of UV light radiation, the electron–hole pairs are generated on the catalyst surface. As a result, the photogenerated electrons over TiO2 are transferred to the possible d-orbit vacant of metals that are present in MMT galleries and/or charges are likely to directly participate in reduction and oxidation processes.49 The positive role of MMT can be attributed to the ability of metals to trap electrons in the conduction band (CB) of TiO2 and the inhibition of electron–hole pair recombination. In this way, photo-excited electrons are separated from holes in the valence band (VB) of the catalyst. Generally, only a very small part of the photogenerated electrons and holes have the chance to migrate from the inside bulk to the surface of photocatalyst and react with water and O2 to produce ˙OH groups and O2˙− groups, respectively. The other parts of electrons and holes are recombined together inside the photocatalyst, resulting in the generation of heat. Then the photo-induced electrons excited to the CB can be taken up by the metals of MMT, thereby reducing the recombination of the photogenerated electron–hole pairs. After getting electrons from MMT, ozone may become a source of hydroxyl radicals (˙OH). Then, both holes and hydroxyl radicals can host the strong oxidation power and degrade CIP efficiently.17 Besides the dominantly-functioned ˙OH, a small amount of reactive atomic oxygen O(1D) is formed in the photocatalytic ozonation process through the photolysis of ozone (eqn (11)–(13)); this also plays a small part in the degradation of the target pollutant by the photocatalytic ozonation system.80 Based on the above, the synergistic effects between photocatalysis and ozonation can be effectively promoted. Consequently, the enhanced degradation of CIP is achieved. Thus, it can be inferred that the TiO2/MMT nanocomposite displays an excellent photocatalytic oxidation activity under UV light irradiation.
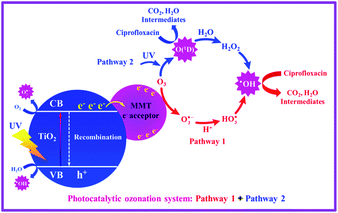 |
| | Fig. 9 Possible mechanism for the degradation of CIP in the photocatalytic ozonation process. | |
3.9. Evaluating the performance of the photocatalytic ozonation process in different real water samples
There are large numbers of inorganic components in real-world water samples that affect the degradation efficiency of the photocatalytic ozonation process. Though TiO2/MMT nanocomposite exhibits high degradation effects on CIP in the photocatalytic ozonation process using deionized water, its performance in different water matrices has to be gauged, because that can be useful for TiO2/MMT application on a larger, technological scale. Hence, to evaluate the capability of the photocatalytic ozonation process in the degradation of CIP, two different types of water (well water and ground water) were used. The main chemical and physical composition features of these waters can be seen in Table 6. Table S4† shows the CIP degradation rate constants obtained using deionized, well, and ground water samples. As shown, in comparison with deionized water, the degradation efficiency of CIP is restrained in well water and ground water samples. The degradation efficiency of the heterogeneous photocatalytic ozonation process decreased from 90.00% with deionized water, to 80.02% with well water, and to 69.99% with ground water. Therefore, the observed decrease in the degradation efficiency of CIP is likely to be a consequence of the presence of inorganic ions in the well and ground water matrices.6,81 The lower k value and degradation efficiency for ground water, as compared to well water, can be due to the high amount of the organic matter that acts as a radiation filter, thereby hindering the production of ˙OH radicals; this, in turn, decreases the degradation rate constant. Moreover, ˙OH radicals can be scavenged by the high concentration of bicarbonate and carbonates present in ground water, forming their respective radicals, which are lower than the oxidation potential of ˙OH radicals.6,73 Hence, the presence of several typical inorganic ions in water matrices subjected to TiO2/MMT photocatalytic ozonation treatment may be a key factor in the successful implementation of TiO2/MMT.
Table 6 Characteristics of well water and ground water samplesa
| Parameters |
Units |
Sample |
| Well water |
Ground water |
| TDS: total dissolved solids, COD: chemical oxygen demand. |
| pH |
— |
7.82 |
7.22 |
| Conductivity |
μS cm−1 |
323 |
117 |
| TDS |
mg L−1 |
226.1 |
90 |
| COD |
mg L−1 |
0.8 |
3 |
| Cl− |
mg L−1 |
9.83 |
0.8 |
| F− |
mg L−1 |
0.12 |
0.33 |
| SO42− |
mg L−1 |
19.84 |
1.54 |
| NO3− |
mg L−1 |
15.99 |
2.69 |
| NO2− |
mg L−1 |
0.04 |
<0.05 |
| BrO3− |
μg L−1 |
<0.95 |
— |
| Na+ |
mg L−1 |
6.11 |
5.48 |
| NH4+ |
mg L−1 |
<0.15 |
<0.01 |
| Mg2+ |
mg L−1 |
<5 |
3.82 |
| Fe2+ |
μg L−1 |
<5 |
<5 |
| Al3+ |
μg L−1 |
<5 |
30 |
| Total hardness |
mg L−1 |
152 |
250 |
| Calcium hardness |
mg L−1 |
80 |
188 |
| Magnesium hardness |
mg L−1 |
72 |
62 |
3.10. Reusability of the nanocomposite
The reusability of the nanocomposite was a very important factor in determining its practical application. Therefore, the reusability of TiO2/MMT during the photocatalytic ozonation process was evaluated under the same operational conditions (Fig. 10). Prior to each reuse, the entire liquid phase of the previous procedure was removed, and the new solution was added without the use of any catalyst treatment. Fig. 10 shows that there was no obvious change in the degradation efficiency after four cycles, where the degradation efficiency slightly decreased from 90.00% to 82.77%; thereby suggesting that the TiO2/MMT nanocomposite was not destroyed during the process and retained good stability. In addition, the small decrease in composite activity could be due to the loss of a small amount of the photocatalyst in the reuse experiments.
 |
| | Fig. 10 Reusability of the TiO2/MMT nanocomposite within four consecutive experimental runs. Conditions: [catalyst]0 = 0.04 g L−1, [CIP]0 = 20 mg L−1, ozone gas flow rate = 2 L h−1 and reaction time = 30 min. | |
3.11. Spectral changes of CIP during photocatalytic ozonation process
The degradation spectrum of CIP by photocatalytic ozonation during 30 min of reaction time was monitored using absorption spectrophotometry (Fig. S3†). Fig. S3† reveals that an increase in the reaction time period led to increasing the degradation efficiency due to the successive degradation of CIP molecules, mainly by the ˙OH radicals formed during the photocatalytic ozonation process. Fig. S3† shows that the two characteristic peaks at λmax of 316 nm and 276 nm gradually decreased over the reaction time. The observed background of the absorption peak could be due to the presence of catalyst particles in the solution, leading to a slight increase in the absorbance values. To achieve the maximum degradation efficiency in the photocatalytic ozonation process, the maximum degradation time was tested (120 min of the reaction time). The optimum conditions for the maximum degradation efficiency of CIP were at a pH 5, with the initial CIP concentration of 20 mg L−1, an ozone gas flow rate of 2 L h−1, and a catalyst dosage of 0.04 g L−1. The results showed that 30 min of reaction time was sufficient to achieve efficient CIP degradation (90%), as no significant degradation efficiency was gained after that time period. Within 90 min of reaction time, the highest degradation efficiency of CIP was achieved at about 95%. Furthermore, the generated intermediates from CIP degradation were analyzed three times by GC-MS, and the common peaks in three analyses were selected and evaluated based on the commercial standards, and the interpretation of their fragment ions were identified in the mass spectra. The retention times (tR) of peaks and the mass spectra of the identified substances through the photocatalytic ozonation process can be seen in Table 7. Eight intermediates with a high match factor of mass spectrum were successfully identified. Some of the intermediates were not detected due to their quick oxidation into derivatives or the low match factors of the chromatographic peaks.6
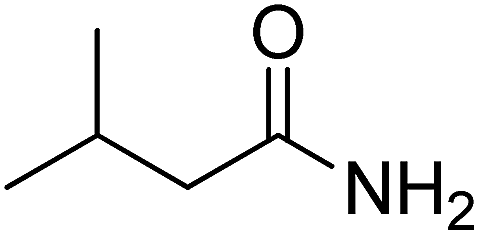
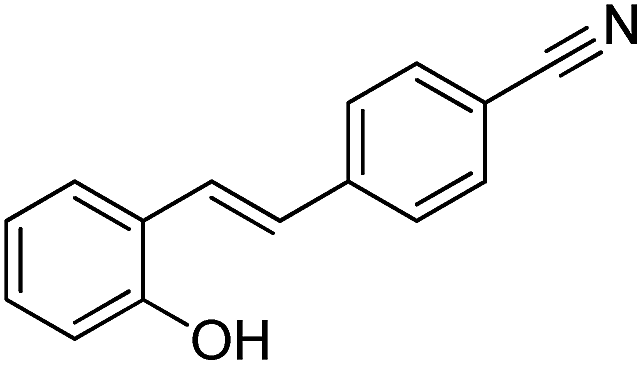
Table 7 Identified by-products during the photocatalytic ozonation of 20 mg L−1 CIP after 15 min of reaction at the ozone flow rate of 2 L h−1 and the pH of 5
| No. |
Compound |
tR (min) |
Main fragments (m/z) (%) |
Structure |
| 1 |
3-Methylbutanamide |
2.77 |
59.00 (100.00%), 75.00 (17.26%), 163.00 (13.92%), 86.10 (11.11%), 133.00 (10.43%) |
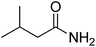 |
| 2 |
(2R,6S)-1,2,6-Trimethylpiperidine |
3.69 |
111.90 (100.00%), 59.00 (64.41%), 75.00 (63.78%), 77.00 (48.30%), 114.00 (27.30%) |
 |
| 3 |
(E)-4-(2-Hydroxystyryl)benzonitrile |
4.08 |
221.00 (100.00%), 75.00 (45.54%), 58.90 (31.96%), 73.00 (26.76%), 222.00 (23.93) |
 |
| 4 |
2,6-Di-tert-butyl-4-methylphenol |
18.02 |
205.10 (100.00%), 220.10 (25.40%), 206.10 (18.27%), 57.00 (10.96%), 145.00 (8.61%) |
 |
| 5 |
3-Methylhenicosane |
29.83 |
57.10 (100.00%), 55.10 (63.94%), 85.10 (52.18%), 71.00 (63.30%), 69.00 (55.71%) |
 |
| 6 |
5-(Cyclohex-1-en-1-yl)-5-ethylpyrimidine-2,4,6(1H,3H,5H)-trione |
34.71 |
207.00 (100.00%), 57.00 (41.72%), 281.00 (38.19%), 55.00 (37.70%), 69.10 (35.65%) |
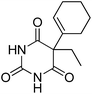 |
| 7 |
3,5-Di-tert-butyl-6-iminocyclohexa-2,4-dienone |
34.88 |
202.00 (100.00%), 57.00 (52.02%), 55.00 (40.88%), 69.10 (39.47%), 281.00 (37.00%) |
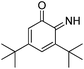 |
| 8 |
4-(1-Methylpiperidin-4-yl)benzene-1,2-diol |
37.69 |
207 (100.00%), 57.00 (65.43%), 69.00 (45.51%), 97.10 (44.40%), 55.10 (43.80%) |
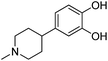 |
4. Conclusion
The degradation of CIP was investigated by different oxidation processes in the presence of TiO2/MMT nanocomposite. TiO2/MMT nanocomposite was synthesized using a simple hydrothermal method. Characterization of the samples by SEM, EDX, HRTEM, FTIR, XRF, N2 adsorption–desorption and UV-vis DRS confirmed the successful synthesis of the samples. Moreover, the effects of the operational parameters, including catalyst dosage, ozone gas flow rate, CIP initial concentration and initial pH on the degradation of CIP were investigated in the photocatalytic ozonation process. Comparing three different CIP degradation processes, including photocatalysis, ozonation and photocatalytic ozonation revealed that photocatalytic ozonation was the most efficient process (degradation efficiency of 90% at 30 min). These results further demonstrated that immobilization of TiO2 on the surface or between the MMT layers produced special electrons and holes transfer from TiO2 to MMT, facilitating the separation of the electron–hole pairs and improving the photocatalytic activity of the nanocomposite during the photocatalytic ozonation process. By using different organic and inorganic radical scavengers, the degradation mechanism of CIP through a photocatalytic ozonation process can be investigated. The results revealed that hydroxyl and superoxide radicals were the main ROSs that were involved in the degradation of CIP. In addition, the importance of catalyst surface reactions such as the adsorption process was confirmed. In addition, a reusability study was performed. The results obtained show the capability of the TiO2/MMT nanocomposite for use in several experimental cycles. Therefore, TiO2/MMT nanocomposite can be an affective photocatalyst for the degradation of organic pollutants under UV light irradiation in a photocatalytic ozonation system. The possible mechanism of CIP degradation in the photocatalytic ozonation process was proposed. GC-MS was also employed to identify some degradation intermediates.
Acknowledgements
The authors thank to TUBITAK for supports by the 2221-Fellowship Program for Visiting Scientists and Scientists on Sabbatical Leave. The financial support by the Atatürk University Scientific Research Project Council (Project No. 2013/308) is gratefully acknowledged. The authors also thank the University of Tabriz (Iran) for all the support.
References
- F. J. Beltrán, A. Aguinaco, J. F. García-Araya and A. Oropesa, Water Res., 2008, 42, 3799–3808 CrossRef PubMed.
- C. Guo, J. Xu, S. Wang, Y. Zhang, Y. He and X. Li, Catal. Sci. Technol., 2013, 3, 1603–1611 CAS.
- M. Fathinia and A. Khataee, Appl. Catal., A, 2015, 491, 136–154 CrossRef CAS.
- F. J. Beltrán, A. Aguinaco and J. F. García-Araya, Appl. Catal., B, 2010, 100, 289–298 CrossRef.
- S. Bae, D. Kim and W. Lee, Appl. Catal., B, 2013, 134–135, 93–102 CrossRef CAS.
- A. Hassani, A. Khataee and S. Karaca, J. Mol. Catal. A: Chem., 2015, 409, 149–161 CrossRef CAS.
- X. Zhang, R. Li, M. Jia, S. Wang, Y. Huang and C. Chen, Chem. Eng. J., 2015, 274, 290–297 CrossRef CAS.
- N. Carmosini and L. S. Lee, Chemosphere, 2009, 77, 813–820 CrossRef CAS PubMed.
- J. E. Renew and C.-H. Huang, J. Chromatogr. A, 2004, 1042, 113–121 CrossRef CAS PubMed.
- K. G. Karthikeyan and M. T. Meyer, Sci. Total Environ., 2006, 361, 196–207 CrossRef CAS PubMed.
- D. G. J. Larsson, C. de Pedro and N. Paxeus, J. Hazard. Mater., 2007, 148, 751–755 CrossRef CAS PubMed.
- W. Shi, Y. Yan and X. Yan, Chem. Eng. J., 2013, 215–216, 740–746 CrossRef CAS.
- P. Huo, Z. Lu, H. Wang, J. Pan, H. Li, X. Wu, W. Huang and Y. Yan, Chem. Eng. J., 2011, 172, 615–622 CrossRef CAS.
- H. Wang, J. Li, P. Huo, Y. Yan and Q. Guan, Appl. Surf. Sci., 2016, 366, 1–8 CrossRef CAS.
- H. Wang, J. Li, C. Ma, Q. Guan, Z. Lu, P. Huo and Y. Yan, Appl. Surf. Sci., 2015, 329, 17–22 CrossRef CAS.
- D. Mao, A. Yu, S. Ding, F. Wang, S. Yang, C. Sun, H. He, Y. Liu and K. Yu, Appl. Surf. Sci., 2016, 389, 742–750 CrossRef CAS.
- Z. Pan, Q. Cai, Q. Luo and X. Li, Synth. React. Inorg. Met.-Org. Chem., 2015, 45, 447–450 CrossRef CAS.
- M. Mehrjouei, S. Müller and D. Möller, J. Photochem. Photobiol., A, 2011, 217, 417–424 CrossRef CAS.
- M. Tahir, B. Tahir and N. S. Amin, Mater. Res. Bull., 2015, 63, 13–23 CrossRef CAS.
- A. R. Khataee and M. B. Kasiri, J. Mol. Catal. A: Chem., 2010, 328, 8–26 CrossRef CAS.
- D. Chen, Q. Zhu, F. Zhou, X. Deng and F. Li, J. Hazard. Mater., 2012, 235–236, 186–193 CrossRef CAS PubMed.
- Y. Li and S.-J. Kim, J. Phys. Chem. B, 2005, 109, 12309–12315 CrossRef CAS PubMed.
- D. Chen, H. Zhu and X. Wang, Appl. Surf. Sci., 2014, 319, 158–166 CrossRef CAS.
- A. Hassani, A. Khataee, S. Karaca and M. Shirzad-Siboni, Environ. Technol., 2015, 36, 3125–3135 CrossRef CAS PubMed.
- M. S. Vohra and K. Tanaka, Water Res., 2003, 37, 3992–3996 CrossRef CAS PubMed.
- A. R. Khataee, M. N. Pons and O. Zahraa, J. Hazard. Mater., 2009, 168, 451–457 CrossRef CAS PubMed.
- S. Anandan and M. Yoon, J. Photochem. Photobiol., C, 2003, 4, 5–18 CrossRef CAS.
- H. Chen, S. W. Lee, T. H. Kim and B. Y. Hur, J. Eur. Ceram. Soc., 2006, 26, 2231–2239 CrossRef CAS.
- A. Hassani, R. Darvishi Cheshmeh Soltani, S. Karaca and A. Khataee, J. Ind. Eng. Chem., 2015, 21, 1197–1207 CrossRef CAS.
- A. Hassani, A. Khataee, S. Karaca, M. Karaca and M. Kıranşan, J. Environ. Chem. Eng., 2015, 3, 2738–2749 CrossRef CAS.
- A. Hassani, R. Darvishi Cheshmeh Soltani, M. Kıranşan, S. Karaca, C. Karaca and A. Khataee, Korean J. Chem. Eng., 2016, 33, 178–188 CrossRef CAS.
- C. Ooka, H. Yoshida, K. Suzuki and T. Hattori, Microporous Mesoporous Mater., 2004, 67, 143–150 CrossRef CAS.
- F. J. Beltrán, A. Aguinaco, A. Rey and J. F. García-Araya, Ind. Eng. Chem. Res., 2012, 51, 4533–4544 CrossRef.
- T. Oyama, T. Otsu, Y. Hidano, T. Koike, N. Serpone and H. Hidaka, Sol. Energy, 2011, 85, 938–944 CrossRef CAS.
- A. Czímerová, J. Bujdák and R. Dohrmann, Appl. Clay Sci., 2006, 34, 2–13 CrossRef.
- H. Bader and J. Hoigné, Water Res., 1981, 15, 449–456 CrossRef CAS.
- Y. Bessekhouad, D. Robert, J. V. Weber and N. Chaoui, J. Photochem. Photobiol., A, 2004, 167, 49–57 CrossRef CAS.
- I. Fatimah, S. Wang and D. Wulandari, Appl. Clay Sci., 2011, 53, 553–560 CrossRef CAS.
- L. Yuan, D. Huang, W. Guo, Q. Yang and J. Yu, Appl. Clay Sci., 2011, 53, 272–278 CrossRef CAS.
- Y. Liu, D. Sun, S. Askari, J. Patel, M. Macias-Montero, S. Mitra, R. Zhang, W.-F. Lin, D. Mariotti and P. Maguire, Sci. Rep., 2015, 5, 15765 CrossRef PubMed.
- S. K. Kansal, S. Sood, A. Umar and S. K. Mehta, J. Alloys Compd., 2013, 581, 392–397 CrossRef CAS.
- A. Hassani, M. Kıranşan, R. Darvishi Cheshmeh Soltani, A. Khataee and S. Karaca, Turk. J. Chem., 2015, 39, 734–749 CrossRef CAS.
- X. Liu, J. Xiong and L. Liang, J. Nat. Gas Sci. Eng., 2015, 22, 62–72 CrossRef CAS.
- B. Tanhaei, A. Ayati, M. Lahtinen and M. Sillanpää, Chem. Eng. J., 2015, 259, 1–10 CrossRef CAS.
- G. Li, J. Lan, J. Liu and G. Jiang, J. Colloid Interface Sci., 2013, 405, 164–170 CrossRef CAS PubMed.
- K. Kočí, L. Matějová, O. Kozák, L. Čapek, V. Valeš, M. Reli, P. Praus, K. Šafářová, A. Kotarba and L. Obalová, Appl. Catal., B, 2014, 158–159, 410–417 CrossRef.
- K. Chen, J. Li, J. Li, Y. Zhang and W. Wang, Colloids Surf., A, 2010, 360, 47–56 CrossRef CAS.
- S. Miao, Z. Liu, B. Han, J. Zhang, X. Yu, J. Du and Z. Sun, J. Mater. Chem., 2006, 16, 579–584 RSC.
- M. Tahir and N. S. Amin, Appl. Catal., B, 2013, 142–143, 512–522 CrossRef CAS.
- F. J. Rivas, F. J. Beltrán, O. Gimeno and M. Carbajo, Ind. Eng. Chem. Res., 2006, 45, 166–174 CrossRef CAS.
- U. Černigoj, U. L. Štangar and P. Trebše, Appl. Catal., B, 2007, 75, 229–238 CrossRef.
- A. Khataee, S. Arefi-Oskoui, M. Fathinia, A. Esmaeili, Y. Hanifehpour, S. W. Joo and N. Hamnabard, J. Mol. Catal. A: Chem., 2015, 398, 255–267 CrossRef CAS.
- S. Wang, F. Shiraishi and K. Nakano, Chem. Eng. J., 2002, 87, 261–271 CrossRef CAS.
- R. Rajeswari and S. Kanmani, Desalination, 2009, 242, 277–285 CrossRef CAS.
- A. Ö. Yıldırım, Ş. Gül, O. Eren and E. Kuşvuran, Clean: Soil, Air, Water, 2011, 39, 795–805 CrossRef.
- L. Xu, H. Zang, Q. Zhang, Y. Chen, Y. Wei, J. Yan and Y. Zhao, Chem. Eng. J., 2013, 232, 174–182 CrossRef CAS.
- F. J. Beltrán, A. Aguinaco and J. F. García-Araya, Water Res., 2009, 43, 1359–1369 CrossRef PubMed.
- M. Mehrjouei, S. Müller and D. Möller, Chem. Eng. J., 2012, 211–212, 353–359 CrossRef CAS.
- A. Aguinaco, F. J. Beltrán, J. F. García-Araya and A. Oropesa, Chem. Eng. J., 2012, 189–190, 275–282 CrossRef CAS.
- M. Fathinia, A. Khataee, S. Aber and A. Naseri, Appl. Catal., B, 2016, 184, 270–284 CrossRef CAS.
- F. J. Beltrán, J. P. Pocostales, P. M. Alvarez and J. Jaramillo, J. Hazard. Mater., 2009, 169, 532–538 CrossRef PubMed.
- F. J. Beltrán, F. J. Rivas and R. Montero-de-Espinosa, Appl. Catal., B, 2002, 39, 221–231 CrossRef.
- S.-N. Zhu, K. N. Hui, X. Hong and K. S. Hui, Chem. Eng. J., 2014, 242, 180–186 CrossRef CAS.
- J. Ge and J. Qu, J. Hazard. Mater., 2003, 100, 197–207 CrossRef CAS PubMed.
- T. A. Gad-Allah, M. E. M. Ali and M. I. Badawy, J. Hazard. Mater., 2011, 186, 751–755 CrossRef CAS PubMed.
- Y. Ma, J. Zhang, B. Tian, F. Chen, S. Bao and M. Anpo, Res. Chem. Intermed., 2012, 38, 1947–1960 CrossRef CAS.
- K. Wang, J. Zhang, L. Lou, S. Yang and Y. Chen, J. Photochem. Photobiol., A, 2004, 165, 201–207 CrossRef CAS.
- M. Sökmen and A. Özkan, J. Photochem. Photobiol., A, 2002, 147, 77–81 CrossRef.
- D. E. Santiago, J. Araña, O. González-Díaz, M. E. Alemán-Dominguez, A. C. Acosta-Dacal, C. Fernandez-Rodríguez, J. Pérez-Peña and J. M. Doña-Rodríguez, Appl. Catal., B, 2014, 156–157, 284–292 CrossRef CAS.
- A. Khataee, A. Khataee, M. Fathinia, Y. Hanifehpour and S. W. Joo, Ind. Eng. Chem. Res., 2013, 52, 13357–13369 CrossRef CAS.
- P.-S. Yap and T.-T. Lim, Appl. Catal., B, 2011, 101, 709–717 CrossRef CAS.
- A.-G. Rincón and C. Pulgarin, Appl. Catal., B, 2004, 51, 283–302 CrossRef.
- C. Sirtori, A. Agüera, W. Gernjak and S. Malato, Water Res., 2010, 44, 2735–2744 CrossRef CAS PubMed.
- L.-S. Zhang, K.-H. Wong, H.-Y. Yip, C. Hu, J. C. Yu, C.-Y. Chan and P.-K. Wong, Environ. Sci. Technol., 2010, 44, 1392–1398 CrossRef CAS PubMed.
- Y. Chen, S. Yang, K. Wang and L. Lou, J. Photochem. Photobiol., A, 2005, 172, 47–54 CrossRef CAS.
- X. Zhang, D. D. Sun, G. Li and Y. Wang, J. Photochem. Photobiol., A, 2008, 199, 311–315 CrossRef CAS.
- J. Rabani, K. Yamashita, K. Ushida, J. Stark and A. Kira, J. Phys. Chem. B, 1998, 102, 1689–1695 CrossRef CAS.
- Y. Liang, S. Lin, L. Liu, J. Hu and W. Cui, Appl. Catal., B, 2015, 164, 192–203 CrossRef CAS.
- Y. Xu, T. Zhou, S. Huang, M. Xie, H. Li, H. Xu, J. Xia and H. Li, RSC Adv., 2015, 5, 41475–41483 RSC.
- J. Xiao, Y. Xie, F. Nawaz, S. Jin, F. Duan, M. Li and H. Cao, Appl. Catal., B, 2016, 181, 420–428 CrossRef CAS.
- M. Sui, S. Xing, L. Sheng, S. Huang and H. Guo, J. Hazard. Mater., 2012, 227–228, 227–236 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra19191f |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.