
Discovery of novel quinazolinones and their acyclic analogues as multi-kinase inhibitors: design, synthesis, SAR analysis and biological evaluation
Abstract
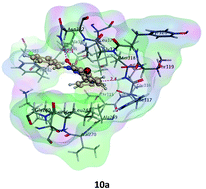
This work deals with the design and synthesis of some novel 6-iodo-2-(pyridin-3/4-yl)-3-substituted quinazolin-4-one derivatives 8a–l, 10a–h, 13–18 in addition to certain acyclic analogues thereof viz. 9a–n and 12a–h. The molecular design strategy was based on structural analogy between the new compounds and reported quinazolines and their acyclic analogues. This design scheme led to the synthesis of 8 new intermediates and 58 new final quinazolinones. The target compounds were evaluated for their antitumor activity against a panel of nine cancer cell lines viz. breast cancer (MCF-7, MDAMB-231, MDAMB-435 and HS-578T), colon cancer (HT-29 and HCC-2998) and leukemia (CCRF-CEM, K-562 and HL-60). The quinazolinones 10a–h displayed exceptional antitumor activity and compounds 12a–h showed superior potency against MCF-7. These compounds were further subjected to in vivo study. Kinase inhibitory assay was also carried out to investigate the mechanism of action of the target compounds and they displayed the highest activity against ABL, ALK and c-RAF kinases. The 3-substituted quinazolinones 10a–h showed the highest kinase activity inhibitory potency against ABL, ALK and c-RAF with the most active compound in this study being the fluoro-3-pyridyl derivative 10a. These results are in compliance with the observed antitumor activity. Finally, a molecular modeling study was performed to interpret the potential molecular interactions of these chemotypes with the most responsive biomolecular target ABL.
 Please wait while we load your content...
Please wait while we load your content...

