
Microwave-assisted hydrothermal synthesis of amorphous MoS2 catalysts and their activities in the hydrodeoxygenation of p-cresol
Kui Wu,
Weiyan Wang*,
Song Tan,
Guohua Zhu,
Liang Tan and
Yunquan Yang*
School of Chemical Engineering, Xiangtan University, Xiangtan, Hunan 411105, PR China. E-mail: wangweiyan@xtu.edu.cn; yangyunquan@xtu.edu.cn
First published on 18th August 2016
Abstract
Based on the normal hydrothermal method, MoS2 amorphous catalysts were synthesized by using molybdenum(V) chloride to replace molybdate under microwave conditions. The catalysts with different structures and morphologies were obtained by changing the synthesis conditions such as pH value, reaction time, temperature and S/Mo molar ratio and their activities were tested in the hydrodeoxygenation (HDO) of p-cresol. The results showed that the structure of MoS2 and its catalytic activity were mainly influenced by the pH value in the synthesis procedure. The surface area decreased with the reduction of pH value. MoS2 possessed a sheet-like shape when the pH was adjusted to an appropriate value. The catalyst synthesis conditions for MoS2 had little effect on the product distribution but affected the conversion in the HDO of p-cresol. The HDO activity of MoS2 depended on the sheet-like shape and slab length. High reaction temperature was beneficial to enhance the deoxygenation degree. The prepared MoS2 had good stability during the reaction, and also presented high activity in the HDO of other phenols such as phenol, o-cresol and 4-ethylphenol. This facile process was easy to operate and the synthesis time for MoS2 was shortened to 0.5 h, which demonstrated its superiority and efficiency.
1. Introduction
At present, the decline of fossil resources and some serious environmental problems make us contribute more and more attention to the development of renewable and sustainable fuels.1 Bio-oil, derived from biomass (such as lignin) by fast pyrolysis, is considered as an alternative for the partial substitution of or supplement for petroleum-based sources.2 However, bio-oil contains many oxygenated compounds, leading its low heating value, poor chemical stability and non-miscibility with petroleum fuels.3 It is necessary to upgrade the bio-oil via reducing its oxygen content. HDO is an effective technology for removing the oxygen by producing water in the presence of hydrogen.4 Among those oxygenated compounds in bio-oil, phenols account for a large proportion and the cleavage of the C–O bond in Ar-OH groups is harder than that in other groups.5 Hence, the HDO of phenols was usually selected as probe to study the activity of the prepared catalysts.Until now, many efforts had been done to improve the HDO activity and deoxygenation degree.5,6 There appeared numbers of HDO catalysts, including normal metals,7–9 noble metals,10–13 metal borides,14–16 sulfides,17–21 carbides22,23 and phosphides.24–26 Noble metals and metal borides exhibited high HDO activities, but they were characterized by high hydrogenation, leading to the saturation of aromatic rings before the cleavage of the carbon–oxygen bond and a high H2 consumption. Furthermore, these catalysts were expensive or unstable at high temperature. Metals, metal carbides and phosphides had low HDO activities, requiring a high reaction temperature or long reaction time to obtain a desired deoxygenation degree. Metal sulfide, a low-cost catalyst, had been widely applied into the HDO of phenols. The deoxygenation degree and product distribution over metal sulfides depended on their morphologies,27 which was closely related to their preparation method.
Generally, Mo-based sulfides are synthesized by impregnation of the corresponding active components over the supports, followed by sulfurization with H2S or CS2–H2.17,18,28 This method contained a sulfurization at high temperature, resulting in some drawbacks such as large particle size and ordered crystalline multi-layered MoS2, which reduced unsaturated S atoms at the edges and then decreased the available active sites for HDO reaction. Recently, defective and amorphous MoS2 with high surface area had been prepared by hydrothermal method using ammonium thiomolybdate (ATTM) as starting material and organic solvent (decalin) as dispersant at 350 °C and showed 2.4-fold higher HDO activity than commercial MoS2.29 K. Faungnawakij et al.30 had reported a new method for the synthesis of Mo-based sulfides with a good HDO activity, i.e., ammonium heptamolybdate and thiourea were mixed together and then heated at 300–450 °C under argon atmosphere. We had also adopted ammonium heptamolybdate and thiourea to prepare low crystalline MoS2 by a hydrothermal method at 200 °C for 12 h.21
Despite current achievements on the synthesis of Mo-based sulfides, how to further simplify the preparation procedure and enhance its HDO activity is still a challenge. The normal hydrothermal method uses electric furnace to transfer thermal energy from the external heat source to the reactants, leading to an inefficient and nonuniform reaction. Moreover, this method spends a long time. Microwave-assisted heating technology assured an internal heating to enhance the reaction rate and avoid the agglomeration phenomenon, which had been developed to be an energy saving, efficient and environmental friendly approach for materials synthesis.31–33 However, to the best of our knowledge, adopting microwave-assisted hydrothermal method to synthesize MoS2 catalyst had not been reported. Therefore, in this study, we reported a fast and simple method to fabricate MoS2 using molybdenum(V) chloride and thiourea as starting materials and water as a solvent. The effects of the preparation conditions such as pH value, reaction time, temperature and S/Mo molar ratio on the structure of MoS2 and their HDO activity were studied in detail.
2. Experimental section
2.1 Catalyst preparation
All solvents and reagents were obtained from Sinopharm Chemical Reagent Co., Ltd. in high purity (≥99%) and used without further purification. MoS2 catalysts were prepared by a facile microwave-assisted hydrothermal method. A typical procedure was shown as following. Molybdenum(V) chloride (2.15 g) and thiourea (1.80 g) were dissolved in 20 mL water and then hydrochloric acid was added into the solution to adjust the pH value to 0.4. Subsequently, the mixture solution was placed in a 100 mL microwave tube and then put into an automated focused microwave system (START SYNTH, milestone) and treated at 200 °C for 30 min. After reaction, the resultant catalysts were separated and washed with water and ethanol for several times. Finally, the resulting product was dried under vacuum at 60 °C for 5 hours. Corresponding names of the samples prepared under different conditions were listed in Table 1.| Samples | pH value | Temperature (°C) | Time (min) | S/Mo ratio | Surface area (m2 g−1) |
|---|---|---|---|---|---|
| Mo–S-1 | 0.7 | 200 | 30 | 3 | 57.0 |
| Mo–S-2 | 0.5 | 200 | 30 | 3 | 40.7 |
| Mo–S-3 | 0.4 | 200 | 30 | 3 | 19.1 |
| Mo–S-4 | 0.3 | 200 | 30 | 3 | 13.0 |
| Mo–S-5 | 0.2 | 200 | 30 | 3 | 13.0 |
| Mo–S-6 | 0.4 | 180 | 30 | 3 | 22.1 |
| Mo–S-7 | 0.4 | 160 | 30 | 3 | 25.6 |
| Mo–S-8 | 0.4 | 200 | 10 | 3 | 20.0 |
| Mo–S-9 | 0.4 | 200 | 15 | 3 | — |
| Mo–S-10 | 0.4 | 200 | 60 | 3 | — |
| Mo–S-11 | 0.4 | 200 | 90 | 3 | 20.0 |
| Mo–S-12 | 0.4 | 200 | 30 | 1 | — |
| Mo–S-13 | 0.4 | 200 | 30 | 2 | 19.3 |
| Mo–S-14 | 0.4 | 200 | 30 | 4 | 21.3 |
2.2 Catalyst characterization
The specific surface area was measured by a Quantachrome's NOVA-2100e Surface Area instrument by physisorption of nitrogen at −196 °C. The samples were dehydrated at 300 °C using vacuum degassing for 12 h before experiments. X-ray diffraction (XRD) measurements were carried on a D/max2550 18 kW Rotating anode X-ray Diffractometer with monochromatic Cu Kα radiation (λ = 1.5418 Å) radiation at voltage and current of 40 kV and 300 mA. The 2θ was scanned over the range of 10–90° at a rate of 10° min−1. The scanning electronic microscopy (SEM) images of the catalysts were obtained on a JEOL JSM-6360 electron microscopy. The morphology was determined by high resolution transmission electron microscopy (HRTEM) on a JEOL JEM-2100 transmission electron microscope with a lattice resolution of 0.19 nm and an accelerating voltage of 200 kV. The samples for the HRTEM study were prepared by the ultrasonic dispersing in ethanol and consequent deposition of the suspension upon a “holey” carbon film supported on a copper grid. The samples were kept under inert atmosphere until the last process.2.3 Catalyst activity measurement
The HDO activity tests were carried out in a 300 mL sealed autoclave. The prepared catalyst (0.3 g) without any further treatment, p-cresol (15.0 g) and dodecane (85.0 g) were placed into the autoclave. Air in the autoclave was evacuated by pressurization–depressurization cycles with nitrogen and subsequently with hydrogen. The system was heated to 300 °C, then pressurized with hydrogen to 4.0 MPa and adjusted the stirring speed to 900 rpm. During the reaction, liquid samples were withdrawn from the reactor and analyzed by Agilent 6890/5973N GC-MS and 7890 gas chromatography using a flame ionization detector (FID) with a 30 m AT-5 capillary column. To separate the reaction products, the temperature in the GC oven was heated from 40 °C to 85 °C with the ramp of 20 °C min−1, held at 85 °C for 4.0 min, then heated to 200 °C at a rate of 20 °C min−1 and kept at 200 °C for 5.0 min. The experiments have been repeated twice at least and the results showed that the conversion and selectivity were within 3.0% of the average values. Conversion is defined as (1 − remained reactant/initial reactant) × 100%; HYD/DDO is defined as total selectivity of methylcyclohexane and 4-methylcyclohexene/the selectivity of toluene; deoxygenation degree (D.D., wt%) is defined as (1 − oxygen content in the final organic compounds/total oxygen content in the initial material) × 100%.3. Results and discussion
3.1 Synthesis of MoS2
Because of the specificity of MoCl5, it reacted violently with water to produce hydrogen chloride and white precipitate. Hence, some acid should be added into the aqueous solution to lower its pH value before dissolving MoCl5. Otherwise, there would produce much Mo oxides and remain very small percentage of Mo5+ for the formation of MoS2. For the hydrothermal synthesis of MoS2 from MoCl5, the occurred reactions were expressed as follows:| NH2SCNH2 + 2H2O → H2S + 2NH3 + CO2 | (1) |
| 2MoCl5 + 5H2S + 10NH3 → 2MoS2 + 10NH4Cl + S | (2) |
The overall reaction was expressed as (3):
| 2MoCl5 + 5NH2SCNH2 + 10H2O → 2MoS2 + 10NH4Cl + S + 5CO2 | (3) |
Obviously, to obtain MoS2, sufficient S2− supply should be ensured, which was closely related to the decomposition of NH2SCNH2. From eqn (1), the formation rate of S2− depended on the pH value, temperature and the added amount of NH2SCNH2, but there also produced basic NH3, which increased the pH value of the system. After optimizing these reaction conditions, it could reveal the structure–activity relationship of the catalyst and obtain MoS2 with high activity for the HDO reaction.
3.2 Catalyst characterization
Fig. 1 shows the XRD patterns of MoS2 prepared under different pH values, reaction temperatures, reaction times and S/Mo molar ratios. The peaks appeared at 14°, 33° and 59° were corresponded to the (0 0 2), (1 0 0) and (1 1 0) planes of MoS2,34,35 respectively. Fig. 1(a) displayed that all the samples prepared at different pH values presented a typical MoS2 structure, but compared with commercial MoS2, the diffraction peaks become significant weaker and broader, which was attributed to the low crystallinity and small size particles. Like reported by B. Yoosuk et al.,29 the prepared MoS2 had an amorphous structure. It had been proposed that the presence of the (1 1 0) peak at 2θ = 59° was representative of a slab layer length.36 With the increase of pH value, the peak intensity ratio of (0 0 2)/(1 0 0) increased firstly and then decreased while the intensity of peak at 59° decreased gradually. In other word, Mo–S-3 presented the largest peak intensity ratio of (0 0 2)/(1 0 0) and Mo–S-4 and Mo–S-5 presented the shorter layer. This relative low (0 0 2)/(1 0 0) peak intensity ratio and the weak peak at 59° were attributed to the defective structure of (0 0 2) plane, which would be directly proved by the TEM characterization results. | ||
| Fig. 1 XRD patterns of MoS2 prepared under different conditions. | ||
As shown in Fig. 1(b), the peak at 14° was obvious, but the peaks to (1 0 0) and (1 1 0) planes decreased with the decrement of reaction temperature, which was mainly attributed to the following reason. The lower temperature decreased the decomposition of NH2SCNH2 and supplied less S2− for the formation of MoS2, consequently, the produced particles were limited, inhibiting the growth along (1 0 0) plane and exhibiting a short slab length. Fig. 1(c) presented that the peak intensity ratio of (0 0 2)/(1 0 0) decreased while the peak at 59° increased when prolonging the reaction time. The intensity decrease of the peak to (0 0 2) plane might be attributed to the loss of S at the edge of MoS2 because of the corrosion at low pH value. This corrosion became more serious with the extension of reaction time. In addition, the longer time the reaction lasted, the more particles aggregated together, and the higher intensity of (1 1 0) peak. For the effect of S/Mo molar ratio on the structure of MoS2, it could be observed from Fig. 1(d) that adding appropriate amount of NH2SCNH2 was beneficial to obtain MoS2 with a good structure. When the S/Mo molar ratio was 1, the created S2− was not sufficient for the formation of MoS2, and MoO3 phase was observed in Mo–S-12.37 In contrast, excessive NH2SCNH2 produced much NH3, which increased the pH value of the reaction system. For example, Mo–S-14 displayed the similar XRD pattern to Mo–S-1.
The prepared MoS2 samples were analysed by HRTEM to study the effect of the reaction conditions on their morphology. As shown in Fig. 2, Mo–S-1 presented some groups of parallel dark thread-like fringes with a distance of 0.65 nm, characterizing the layer edges, which was corresponded to (0 0 2) basal plane of MoS2.35,38 With the decrease of pH value, these fringes became short and unobvious, especial at very low pH value. The average slab length calculated based on many HRTEM images for Mo–S-1, Mo–S-3 and Mo–S-5 was 4.2 nm, 2.9 nm and 1.8 nm, respectively, which were well consistent with the XRD results. The effect of reaction temperature suggested that the low temperature was not favourable for the formation of layer structure and the slab growth, exhibiting an increase of slab length with the reaction temperature. This was ascribed to the slow decomposition of NH2SCNH2 at low temperature, which leaded to the low production of MoS2 and then inhibited its growth. Mo–S-8 displayed some dark fringes with short length, while some long fringes with defective structure were observed in the HRTEM image of Mo–S-11. These suggested that the MoS2 slab gradually grew to long one and the corrosion became more serious under an acidic condition with the increase of reaction time. This destruction of (0 0 2) basal plane gave the evidence for the lower peak intensity ratio of (0 0 2)/(1 0 0) in the XRD of Mo–S-11 than that of Mo–S-3.
 | ||
| Fig. 2 HRTEM images of MoS2 prepared under different conditions. | ||
The pH value also had a great effect on the morphology of MoS2. Fig. 3 displays the SEM images of some MoS2 prepared under different conditions. Mo–S-1 was consisted of small irregular particles. When the pH value decreased to 0.4, the produced MoS2 aggregated together to form spherical particles. Moreover, there presented many sheets with a random self-assembly on the surface of Mo–S-3, which was expected to expose more edge sites for the catalytic reaction. At a very low pH value, the random self-assembly of sheets was inhibited, e.g., Mo–S-5 presented spherical particles with smooth surface. Mo–S-6 and Mo–S-11 presented the similar morphology as Mo–S-3. Brunauer–Emmett–Teller (BET) analysis also supported these results. As shown in Table 1, the surface area of Mo–S-1 was the largest (57.0 m2 g−1), which was decreased with the reduction of pH value. Because of the smooth and large particles, Mo–S-5 had the smallest surface area. However, the other factors such reaction time and S/Mo molar ratio had little effect on the surface area of MoS2.
 | ||
| Fig. 3 SEM images of MoS2 prepared under different conditions. | ||
3.3 HDO activity of MoS2 in the HDO of p-cresol
To study the reaction routes for the HDO of p-cresol on MoS2 catalysts prepared by the microwave-assisted hydrothermal process, the changes of reactant and products concentrations versus time are showed in Fig. 4. The products were 4-methylcyclohexanol, 4-methylcyclohexene, methylcyclohexane and toluene, where toluene was the main product. The appearance of 4-methylcyclohexanol indicated that some of p-cresol was firstly hydrogenated via saturating the benzene ring. It had been proposed that the conversion of toluene into methylcyclohexane on Mo based sulfide catalysts could be neglected because of its very low hydrogenation activity.39 Fig. 4 displayed that both the concentrations of methylcyclohexane and toluene increased with reaction time, indicating that there existed two parallel route in the HDO of p-cresol on Mo–S-3: (i) p-cresol → 4-methylcyclohexanol → 4-methylcyclohexene → methylcyclohexane (HYD) and (ii) p-cresol → toluene (direct deoxygenation, DDO). During the whole HDO reaction, the product concentration decreased in the order of toluene ≫ methylcyclohexane > 4-methylcyclohexene > 4-methylcyclo-hexanol. These demonstrated that the prepared catalyst had high deoxygenation active at 300 °C and the primary reaction route was DDO, which was consistent with the previous investigations.29,40 Rim–Edge model was a good one to explain the reaction mechanism on unsupported MoS2.41 As shown in Fig. 2, parallel dark thread-like fringes were stated as active sites, where the top and bottom discs were defined as rim sites while the discs between the top and bottom discs were defined as edge sites, which were corresponded to hydrogenation and hydrogenolysis, respectively. Hence, these dark fringes would have a great effect on the HDO activity. | ||
| Fig. 4 Concentrations of reactant and products versus reaction time in the HDO of p-cresol on Mo–S-3 at 300 °C. | ||
The activity comparison of MoS2 prepared under different pH values and temperatures in the HDO of p-cresol are shown in Table 2. After reaction at 300 °C for 6 h, the conversion of p-cresol on Mo–S-1 was 83.8% with a selectivity of 89.0% toluene. With the reduction of pH value for the synthesis of MoS2, the conversion increased firstly and then decreased, but the products distribution changed little. Mo–S-3 exhibited the highest HDO activity: 95.4% conversion with a deoxygenation degree of 92.6%. Considering the surface areas of Mo–S-2 and Mo–S-3, Table 2 showed that the HDO activity of MoS2 increased with the decrease of surface area. However, the conversion and deoxygenation degree on Mo–S-5 with a surface area of 13.0 m2 g−1 was decreased to 54.8% and 50.7%, respectively. Generally, the catalyst with higher surface area would expose more available active sites and enhance its catalytic activity. In this case, it was hard to determinate the relation between surface area and HDO activity. For comparison, MoS2 was also prepared by the normal hydrothermal method (named as Mo–S-3-N) under the same conditions as that for Mo–S-3-N, except removing the microwave environment and prolonging the reaction time to 24 h. The conversion and deoxygenation degree on Mo–S-3-N was 92.2% and 89.7%, respectively, as shown in Table 2, both of which were lower than that on Mo–S-3. But the prepared time for Mo–S-3-N was 48-fold more than that for Mo–S-3. These demonstrated the superiority and high efficiency of microwave-assisted hydrothermal method.
| Catalyst | Mo–S-1 | Mo–S-2 | Mo–S-3 | Mo–S-4 | Mo–S-5 | Mo–S-6 | Mo–S-7 | Mo–S-3-Na |
|---|---|---|---|---|---|---|---|---|
| a The preparation conditions were pH = 0.4, temperature = 200 °C, S/Mo ratio = 3, time = 24 h and without microwave radiation. | ||||||||
| Conversion | 83.8 | 87.8 | 95.4 | 93.1 | 54.8 | 84.0 | 75.6 | 92.2 |
| Product distribution | ||||||||
| 4-Methylcyclohexanol | 1.4 | 1.3 | 1.9 | 1.1 | 0.4 | 1.8 | 1.0 | 1.3 |
| 4-Methylcyclohexene | 2.6 | 3.5 | 3.3 | 3.3 | 3.7 | 5.4 | 5.1 | 3.4 |
| Methylcyclohexane | 6.8 | 8.0 | 9.0 | 7.2 | 6.6 | 10.0 | 7.2 | 10.7 |
| Toluene | 89.0 | 87.2 | 85.7 | 88.5 | 89.3 | 82.8 | 86.8 | 84.6 |
| HYD/DDO | 0.12 | 0.15 | 0.17 | 0.13 | 0.12 | 0.21 | 0.15 | 0.18 |
| Deoxygenation degree | 80.3 | 84.8 | 92.6 | 90.9 | 50. 7 | 80.2 | 71.9 | 89.7 |
However, MoS2 was a structure-sensitive catalyst. Its structure was also a significant factor for its catalytic activity. As shown in Fig. 3, the morphology of Mo–S-1, Mo–S-3 and Mo–S-5 presented small irregular particles, large particles composed sheets and large particles with smooth surface, respectively. Moreover, the average slab length of Mo–S-5 was decreased to 1.8 nm, which might be unfavourable for the adsorption of reactant molecules. The HDO of p-cresol on MoS2 prepared under different temperature also presented this similar phenomenon. At low temperature, e.g., 160 °C, the (0 0 2) plane of MoS2 structure was not formed completely. Although some black fringes were observed in the HRTEM image of Mo–S-6, the average slab length was very short. Table 2 showed that the conversion on Mo–S-6 and Mo–S-7 was 11.4% and 19.8% lower than that on Mo–S-3, respectively, indicating that the defective (0 0 2) plane structure and short slab length inhibited the adsorption of p-cresol molecules, which resulted in a low conversion. These suggested that the morphology had greater effect for the catalytic activity of MoS2 than the surface area and the sheet shape exhibited the highest activity. According to the Rim–Edge model, this sheet shape exposed more active sites at its edges for the HDO reactions and then contributed to the higher HDO activity.
Table 3 shows the activity of MoS2 prepared under different time and S/Mo molar ratios in the HDO of p-cresol. Previous studies had proposed that HYD and DDO started by flat η5 adsorption through aromatic ring and the vertical η1 adsorption of phenols through oxygen on all the edges of MoS2,17,41 respectively. According to the conversions and products distribution in Table 3, it could concluded that the catalyst preparation conditions only influenced the adsorption amount of reactant but not to adsorption models. The deoxygenation degree increased with the orders of Mo–S-8 (69.4%) < Mo–S-9 (77.1%) < Mo–S-11 (83.6%) < Mo–S-10 (88.7%) < Mo–S-3 (92.6%) and Mo–S-14 (88.0%) ≈ Mo–S-12 (88.3%) < Mo–S-3 (92.6%) ≈ Mo–S-13 (92.8%). This difference for MoS2 on the HDO activity was mainly resulted from its morphology because these catalysts almost possessed the same surface area. As shown in Fig. 2, the (0 0 2) plane structure for Mo–S-8 was not very obvious, similarly to the catalysts prepared under different pH values and temperatures, leading a reduced adsorption of p-cresol on Mo–S-8 compared with that on Mo–S-3. According to coordinatively unsaturated sites theory that the elimination of sulfur atom on the edges of the MoS2 slabs created active sites for the reactions, Mo–S-11 should presented higher HDO activity than Mo–S-3. However, the sulfur–oxygen exchange was the reason for the deactivation of Mo based sulfides.42 MoS2 was continuously converted into Mo oxide during the HDO reaction, leading to the reduction of deoxygenation degree. The lower conversion on Mo–S-12 than that on Mo–S-3 was mainly resulted from the appearance of MoO3 phase because MoS2 had higher HDO activity than MoO3.37 The HDO activity results of MoS2 prepared by adjusting the S/Mo ratio further verified the effect of pH value. When adding excessive NH2SCNH2, e.g., for Mo–S-14, its decomposition increased the pH value of the reaction system, and its HDO activity was between Mo–S-2 and Mo–S-3.
| Catalyst | Mo–S-8 | Mo–S-9 | Mo–S-10 | Mo–S-11 | Mo–S-12 | Mo–S-13 | Mo–S-14 |
|---|---|---|---|---|---|---|---|
| Conversion | 73.2 | 80.5 | 91.2 | 86.8 | 90.5 | 95.3 | 91.0 |
| Product distribution | |||||||
| 4-Methylcyclohexanol | 0.8 | 1.0 | 1.2 | 1.4 | 0.8 | 1.6 | 1.6 |
| 4-Methylcyclohexene | 4.7 | 3.8 | 3.9 | 3.7 | 6.0 | 6.3 | 3.3 |
| Methylcyclohexane | 6.1 | 5.8 | 9.8 | 9.3 | 5.2 | 7.1 | 8.4 |
| Toluene | 88.3 | 89.4 | 85.1 | 85.6 | 88.0 | 85.1 | 86.7 |
| HYD/DDO | 0.13 | 0.12 | 0.17 | 0.17 | 0.15 | 0.14 | 0.18 |
| Deoxygenation degree | 69.4 | 77.1 | 88.7 | 83.6 | 88.3 | 92.8 | 88.0 |
Reaction temperature was a crucial factor for the conversion and product distribution in the HDO of p-cresol. As shown in Fig. 5, after reaction on Mo–S-3 at 275 °C for 6 h, the conversion was 67.7% with a selectivity of 85.3% toluene, and the corresponding deoxygenation degree and HYD/DDO was 63.7% and 0.17, respectively. Oxygen-containing compound selectivity was only 0.7%, indicating that this catalyst had a high deoxygenation activity. When the temperature increased to 325 °C, the total selectivity of 4-methylcyclohexene and methylcyclohexane decreased to 6.5% while toluene selectivity increased to 92.0%. The calculated HYD/DDO decreased to 0.09. Higher temperature was favorable to activate the C–O bond in p-cresol and lowered the hydrogen concentration in organic solvents,20 resulting in the increase on the conversion, deoxygenation degree and DDO route, which was consistent with previous investigations.37,43,44
 | ||
| Fig. 5 HDO of p-cresol on Mo–S-3 at different reaction temperatures for 6 h. | ||
Mo–S-3, the optimized catalyst, was also applied into the HDO of phenol, o-cresol and 4-ethylphenol. As shown in Fig. 6, after reaction at 300 °C for 6 h, phenol, o-cresol and 4-ethylphenol conversion was 84.0%, 73.0% and 92.5%, respectively. Compared with the HDO of p-cresol, both the conversions of phenol and o-cresol were decreased, which were attributed to the electronic effect and steric effect of substituent groups. For these three reactions, the oxygen-containing products selectivity in each HDO reaction was very low, while the aromatics selectivity was higher than 88%, which further verified the dominant reaction route of DDO and a high activity in the HDO of phenols.
 | ||
| Fig. 6 HDO of phenol, o-cresol and 4-ethylphenol on Mo–S-3 at 300 °C for 6 h. | ||
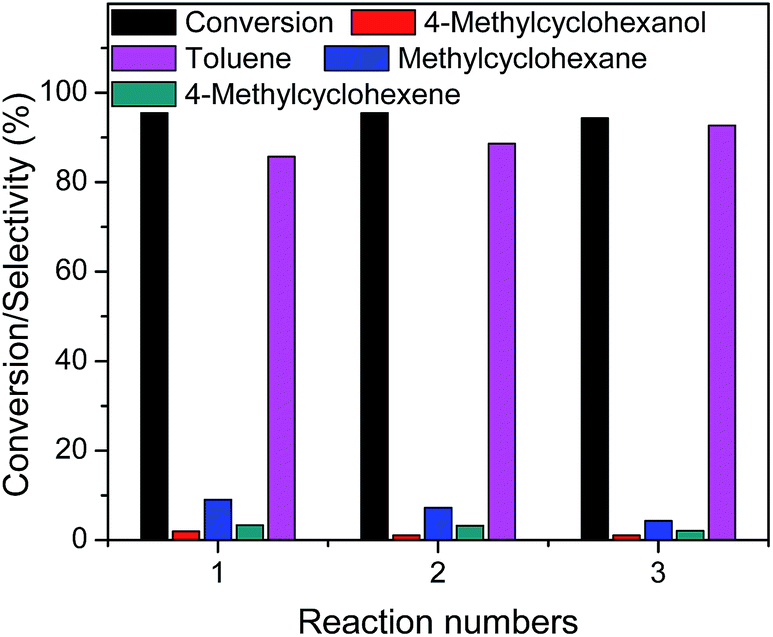
Mo–S-3 was also selected to study the reusability of the synthesized catalyst in the HDO of p-cresol. The results are presented in Fig. 7. After reaction at 300 °C for 6 h for three times, the conversion was still up to 94.3%, which was only 1.1% lower than that in the first reaction.42 However, the direct deoxygenation product (toluene) selectivity increased with reaction times. The HYD/DDO decreased to 0.08 after reaction for 3 times. This may be related to the modification of water on the catalyst surface. Previous investigation45 had also reported that the number or strength of acid sites were increased after the adsorption of water on the catalyst surface and thereby enhance hydrogenolysis activity. These indicated that MoS2 catalyst surface was modified by water during the HDO of p-cresol, which enhanced the DDO route, but this promotion mechanism needs to be further studied.
 | ||
| Fig. 7 The reusability of Mo–S-3 in the HDO of p-cresol. | ||
There had developed several method for the synthesis of MoS2 and the catalysts presented different activity. For example, K. Smith et al.27 had compared the HDO activity of MoS2 prepared by different methods based on the reaction rate constant and found that exfoliated MoS2 fabricated by intercalating n-butyllithium into MoS2 under argon atmosphere in a glove box exhibited the highest HDO activity: p-cresol conversion and toluene selectivity were 75% and 36% after reaction at 350 °C for 7 h when the p-cresol/catalyst weight ratio was fixed to 14.4, respectively. Recently, because of the synergy between W and Mo, D. Wang et al.40 had adopted mechanical activation method to prepare W–Mo sulfide catalyst at 600 °C and reported a conversion of 50.0% with a selectivity of 89.0% toluene in the HDO of p-cresol at 300 °C for 5 h when the p-cresol/catalyst weight ratio was fixed to 13.9. More recently, we had synthesized MoS2 nano-sheets by hydrothermal method in a quartz reactor at 200 °C for 12 h and obtained a deoxygenation degree of 99.9% in the HDO of p-cresol at 300 °C for 4 h when the p-cresol/catalyst weight ratio was fixed to 22.5.21 However, in this study, the preparation time for MoS2 by microwave-assisted hydrothermal method was shortened to 0.5 h at 200 °C. Moreover, the prepared MoS2 presented high activity in the HDO of p-cresol: 92.6% deoxygenation degree at 300 °C for 6 h. In addition, this method was easy to operate.
4. Conclusion
In this study, MoS2 was synthesized by microwave-assisted hydrothermal method using molybdenum(V) chloride and thiourea as starting materials, where the required reaction time was markedly shortened. The pH value of the reaction system had a great effect on the surface area and morphology of MoS2. After optimizing the synthesis conditions such as pH value, reaction time, temperature and S/Mo molar ratio in the initial solution, MoS2 presented high activity in the HDO of p-cresol: the conversion and deoxygenation degree reached to 95.4% and 92.4% at 300 °C for 6 h, respectively. MoS2 with defective of (0 0 2) plane structure and very short slab length exhibited low activity. The higher HDO activity of MoS2 was attributed to that its sheet morphology could expose more active sites for the reaction. Higher temperature was beneficial to increase the deoxygenation degree. The prepared MoS2 also exhibited high activity in the HDO of other phenols and good stability. Compared with the first reaction, the conversion was only decreased by 1.1% after reaction for three times, but the toluene selectivity was increased with reaction times. More importantly, this microwave-assisted hydrothermal method was easy to operate and saved much time, demonstrating its superiority and high efficiency, which could also be applied for the synthesis of Ni/Co promoted Mo based sulfides and other sulfides with high catalytic activity.Acknowledgements
This research was supported by the National Natural Science Foundation of China (No. 21306159, 21376202), Scientific Research Fund of Hunan Provincial Education Department (15B234) and National Students' innovation and entrepreneurship training program (201510530008).Notes and references
- Z. Liu, D. Guan, W. Wei, S. J. Davis, P. Ciais, J. Bai, S. Peng, Q. Zhang, K. Hubacek, G. Marland, R. J. Andres, D. Crawford-Brown, J. Lin, H. Zhao, C. Hong, T. A. Boden, K. Feng, G. P. Peters, F. Xi, J. Liu, Y. Li, Y. Zhao, N. Zeng and K. He, Nature, 2015, 524, 335–338 CrossRef CAS PubMed.
- C. Li, X. Zhao, A. Wang, G. W. Huber and T. Zhang, Chem. Rev., 2015, 115, 11559–11624 CrossRef CAS PubMed.
- C. Xu, R. A. D. Arancon, J. Labidi and R. Luque, Chem. Soc. Rev., 2014, 43, 7485–7500 RSC.
- H. Wang, J. Male and Y. Wang, ACS Catal., 2013, 3, 1047–1070 CrossRef CAS.
- H. Shafaghat, P. S. Rezaei and W. M. Ashri Wan Daud, RSC Adv., 2015, 5, 103999–104042 RSC.
- M. Saidi, F. Samimi, D. Karimipourfard, T. Nimmanwudipong, B. C. Gates and M. R. Rahimpour, Energy Environ. Sci., 2014, 7, 103–129 CAS.
- Y. Yang, C. Ochoa-Hernández, V. A. de la Peña O'Shea, P. Pizarro, J. M. Coronado and D. P. Serrano, Appl. Catal., B, 2014, 145, 91–100 CrossRef CAS.
- R. G. Kukushkin, O. A. Bulavchenko, V. V. Kaichev and V. A. Yakovlev, Appl. Catal., B, 2015, 163, 531–538 CrossRef CAS.
- A. B. Dongil, I. T. Ghampson, R. Garcia, J. L. G. Fierro and N. Escalona, RSC Adv., 2016, 6, 2611–2623 RSC.
- C. Zhao, Y. Kou, A. A. Lemonidou, X. Li and J. A. Lercher, Angew. Chem., Int. Ed., 2009, 48, 3987–3990 CrossRef CAS PubMed.
- Y.-K. Hong, D.-W. Lee, H.-J. Eom and K.-Y. Lee, Appl. Catal., B, 2014, 150–151, 438–445 CrossRef CAS.
- Q. Tan, G. Wang, L. Nie, A. Dinse, C. Buda, J. Shabaker and D. E. Resasco, ACS Catal., 2015, 5, 6271–6283 CrossRef CAS.
- K. L. Luska, P. Migowski, S. El Sayed and W. Leitner, Angew. Chem., Int. Ed., 2015, 54, 15750–15755 CrossRef CAS PubMed.
- W. Wang, Z. Qiao, K. Zhang, P. Liu, Y. Yang and K. Wu, RSC Adv., 2014, 4, 37288–37295 RSC.
- W. Wang, S. Yang, Z. Qiao, P. Liu, K. Wu and Y. Yang, Catal. Commun., 2015, 60, 50–54 CrossRef CAS.
- W. Wang, P. Liu, K. Wu, S. Tan, W. Li and Y. Yang, Green Chem., 2016, 18, 984–988 RSC.
- Y. Romero, F. Richard and S. Brunet, Appl. Catal., B, 2010, 98, 213–223 CrossRef CAS.
- V. N. Bui, D. Laurenti, P. Afanasiev and C. Geantet, Appl. Catal., B, 2011, 101, 239–245 CrossRef CAS.
- S. Mukundan, M. Konarova, L. Atanda, Q. Ma and J. Beltramini, Catal. Sci. Technol., 2015, 5, 4422–4432 CAS.
- W. Wang, L. Li, K. Wu, K. Zhang, J. Jie and Y. Yang, Appl. Catal., A, 2015, 495, 8–16 CrossRef CAS.
- W. Wang, L. Li, K. Wu, G. Zhu, S. Tan, W. Li and Y. Yang, RSC Adv., 2015, 5, 61799–61807 RSC.
- P. M. Mortensen, H. W. P. de Carvalho, J.-D. Grunwaldt, P. A. Jensen and A. D. Jensen, J. Catal., 2015, 328, 208–215 CrossRef CAS.
- C.-J. Chen, W.-S. Lee and A. Bhan, Appl. Catal., A, 2016, 510, 42–48 CrossRef CAS.
- Y. Yang, C. Ochoa-Hernández, P. Pizarro, V. A. de la Peña O'Shea, J. M. Coronado and D. P. Serrano, Fuel, 2015, 144, 60–70 CrossRef CAS.
- Y. Li, X. Yang, L. Zhu, H. Zhang and B. Chen, RSC Adv., 2015, 5, 80388–80396 RSC.
- M. Peroni, G. Mancino, E. Baráth, O. Y. Gutiérrez and J. A. Lercher, Appl. Catal., B, 2016, 180, 301–311 CrossRef CAS.
- Y. Q. Yang, C. T. Tye and K. J. Smith, Catal. Commun., 2008, 9, 1364–1368 CrossRef CAS.
- M. Grilc, B. Likozar and J. Levec, Appl. Catal., B, 2014, 150–151, 275–287 CrossRef CAS.
- B. Yoosuk, D. Tumnantong and P. Prasassarakich, Fuel, 2012, 91, 246–252 CrossRef CAS.
- V. Itthibenchapong, C. Ratanatawanate, M. Oura and K. Faungnawakij, Catal. Commun., 2015, 68, 31–35 CrossRef CAS.
- Y.-J. Zhu and F. Chen, Chem. Rev., 2014, 114, 6462–6555 CrossRef CAS PubMed.
- A. Phuruangrat, D. J. Ham, S. J. Hong, S. Thongtem and J. S. Lee, J. Mater. Chem., 2010, 20, 1683–1690 RSC.
- D. H. Youn, C. Jo, J. Y. Kim, J. Lee and J. S. Lee, J. Power Sources, 2015, 295, 228–234 CrossRef CAS.
- Z. Wu, B. Li, Y. Xue, J. Li, Y. Zhang and F. Gao, J. Mater. Chem. A, 2015, 3, 19445–19454 CAS.
- S. Muralikrishna, K. Manjunath, D. Samrat, V. Reddy, T. Ramakrishnappa and D. H. Nagaraju, RSC Adv., 2015, 5, 89389–89396 RSC.
- Y. Yi, B. Zhang, X. Jin, L. Wang, C. T. Williams, G. Xiong, D. Su and C. Liang, J. Mol. Catal. A: Chem., 2011, 351, 120–127 CrossRef CAS.
- V. M. L. Whiffen and K. J. Smith, Energy Fuels, 2010, 24, 4728–4737 CrossRef CAS.
- Y. Lu, X. Yao, J. Yin, G. Peng, P. Cui and X. Xu, RSC Adv., 2015, 5, 7938–7943 RSC.
- O. I. Şenol, E. M. Ryymin, T. R. Viljava and A. O. I. Krause, J. Mol. Catal. A: Chem., 2007, 277, 107–112 CrossRef.
- C. Wang, Z. Wu, C. Tang, L. Li and D. Wang, Catal. Commun., 2013, 32, 76–80 CrossRef CAS.
- M. Daage and R. R. Chianelli, J. Catal., 1994, 149, 414–427 CrossRef CAS.
- M. Badawi, J. F. Paul, S. Cristol, E. Payen, Y. Romero, F. Richard, S. Brunet, D. Lambert, X. Portier, A. Popov, E. Kondratieva, J. M. Goupil, J. El Fallah, J. P. Gilson, L. Mariey, A. Travert and F. Maugé, J. Catal., 2011, 282, 155–164 CrossRef CAS.
- V. M. L. Whiffen, K. J. Smith and S. K. Straus, Appl. Catal., A, 2012, 419–420, 111–125 CrossRef CAS.
- Y. Yang, H. A. Luo, G. Tong, K. J. Smith and C. T. Tye, Chin. J. Chem. Eng., 2008, 16, 733–739 CrossRef CAS.
- C. N. Satterfield, C. M. Smith and M. Ingalls, Ind. Eng. Chem. Process Des. Dev., 1985, 24, 1000–1004 CAS.
| This journal is © The Royal Society of Chemistry 2016 |