
Synthesis of nanosphere TiO2 with flower-like micro-composition and its application for the selective catalytic reduction of NO with NH3 at low temperature
Hong Wanga,
Kasha Caia,
Jixing Liub,
Xiangjun Zhanga,
Yan Lia,
Kai Chengb,
Jian Liu*b,
Cuiqing Lia,
Fuchen Dinga and
Yongji Songa
aDepartment Chemical Engineering, Beijing Institute of Petrochemical Technology, Beijing, 102617, China
bState Key Laboratory of Heavy Oil Processing, China University of Petroleum, Beijing, 102249, China. E-mail: liujian@cup.edu.cn
First published on 31st August 2016
Abstract
TiO2 nanospheres consisting of flower-like nanopowders were synthesized by a solvothermal method, and Cu/TiO2(T) catalysts were prepared via an impregnation method. Their catalytic performances for the selective catalytic reduction of nitric oxide (NO) with ammonia (NH3-SCR) were investigated. Their structures, morphology and surface components were characterized via X-ray diffraction (XRD), Raman spectroscopy (Raman), scanning electron microscopy (SEM), N2 adsorption–desorption isotherms, X-ray photoelectron spectroscopy (XPS), temperature-programmed desorption of NO (NO-TPD) or O2 (O2-TPD) and temperature-programmed reduction of H2 (H2-TPR) or CO (CO-TPR). The largest specific surface area (236.78 m2 g−1) is obtained for nanosphere TiO2 with flower-like morphology. XPS results show that there are CuO and Cu2O species in Cu(3)/TiO2(T) catalysts. The Cu(3)/TiO2(160) catalyst exhibits the best activity, and its T95 temperature is 170 °C, and the temperature window of NO conversion over 95% is from 170 °C to 310 °C. This is due to the excellent redox properties and the adsorption properties of the catalyst. In situ DRIFTS results demonstrate that Lewis acid sites are involved in NH3-SCR reaction and the adsorption and activation of NH3 play a key role in the process of NH3-SCR over Cu/TiO2(T) catalysts.
1. Introduction
Nitrogen oxides (NOx), emitted into the atmosphere from stationary and mobile sources, are considered to be harmful to ecosystems and human beings. It is urgent to eliminate NOx from flue gas for protection of the environment. Selective catalytic reduction (SCR) of NOx with NH3 is currently considered as the most efficient technology for NOx removal from stationary sources.1–4 The catalyst is the key factor to determine the catalytic efficiency. Titania-supported V2O5 catalysts exhibit their optimum performance in a narrow temperature window (300–400 °C) for NH3-SCR. However, a low catalytic activity is observed below 200 °C. Therefore, it is important to develop catalysts that exhibit good activity at low temperature for application in denitrification technology.NH3-SCR includes three main reactions depending on the NO2/NOx ratio in the feed.1 The “standard SCR” reaction, the nitric oxide (NO) is the major NOx component (>90%), refers to the reaction between NO and NH3:
| 4NH3 + NO + O2 → 4N2 + 6H2O | (1) |
The “fast SCR” reaction, where NO2 and NO are in equimolar concentrations in the feed, is defined as:
| 2NH3 + NO + NO2 → 2N2 + 3H2O | (2) |
The “slow SCR” or “NO2-SCR” reaction between NO2 and NH3, the concentration of NO2 is larger than NO, is defined as:
| 4NH3 + NO2 → 3.5N2 + 6H2O | (3) |
In general, the “standard SCR” is the main reaction in the process of selective catalytic reduction of nitrogen oxides. Transition metal oxides show good NH3-SCR activity in the low temperature range. Particularly, copper-based catalysts have attracted much interest owing to their unique redox properties, which make them useful for a variety of applications. The copper-based catalysts for the selective catalytic reduction of NO with NH3 at low temperatures, such as CuOx/WOx–ZrO2,2,4 CuO–CeO2,3,5 Cu/ZSM-5,6–9 Cu-SAPO-34,1,8,10 Cu-SSZ-13,8,11–14 Cu/BEA,15 Cu/TiO2,16–18 and Cu/AC,19,20 have been studied. It has been observed that the carrier has a large influence on the performance of the catalyst. Even the traditional TiO2 carrier for NH3-SCR catalysts, different sources and surface areas will show very different catalytic activities.21 TiO2 carriers synthesized by different methods with different materials exhibit widely different morphologies, as well as physical and chemical properties.22–25 In general, the surface area of TiO2 is lower than 100 m2 g−1. A carrier with a large surface area is beneficial to that the activity components are highly dispersed on the carrier. It not only reduces the amount of active components, but also improves its catalytic performance.
Herein, TiO2 nanospheres with high surface areas were synthesized by the solvothermal method. Cu/TiO2 catalysts were prepared by the impregnation method for NH3-SCR reaction. The effects of TiO2 morphologies on the catalytic performance were investigated.
2. Experimental
2.1 Material preparation
TiO2 nanospheres were synthesized by the solvothermal method described previously.23,24 In a typical procedure, tetrabutyl titanate (TBT) was added dropwise to acetic acid (HAc), with a HAc/TBT volume ratio = 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, with continuous stirring. The resultant mixture was subsequently transferred to a 50 mL stainless steel autoclave with a Teflon inner liner, which was kept at 140 °C, 150 °C, 160 °C or 170 °C for 12 h. After cooling to room temperature, the powder product was separated by centrifugation, washed with distilled water and ethyl-alcohol for three times to remove impurities, then dried at 110 °C for 4 h before being calcined in a tubular oven at 400 °C for 30 min under continuous air flow (400 mL min−1). Finally, white TiO2 powder was obtained.
:1, with continuous stirring. The resultant mixture was subsequently transferred to a 50 mL stainless steel autoclave with a Teflon inner liner, which was kept at 140 °C, 150 °C, 160 °C or 170 °C for 12 h. After cooling to room temperature, the powder product was separated by centrifugation, washed with distilled water and ethyl-alcohol for three times to remove impurities, then dried at 110 °C for 4 h before being calcined in a tubular oven at 400 °C for 30 min under continuous air flow (400 mL min−1). Finally, white TiO2 powder was obtained.
A series of Cu/TiO2 catalysts were prepared by the impregnation method. The loading amounts of copper were 3 wt% with TiO2 as the basis. The required amounts of TiO2 support were added to a beaker containing certain amounts of copper nitrate in deionized water with continuous stirring for 4 hours at room temperature. The excess water was then evaporated on a rotary evaporator at 70 °C. The obtained materials were calcined in a tubular oven at 400 °C for 1 h under continuous air flow (400 mL min−1). The catalysts are labeled as Cu(3)/TiO2(T), where the T given in parentheses is the crystallization temperature of TiO2 synthesis. A series of Cu(x)/TiO2(160) catalysts with different Cu loading amounts (x = 1, 5, 7 and 9) were also prepared by the impregnation method. The state-of-the-art SCR catalyst 3 wt% V2O5-9 wt% WO3/TiO2 was also prepared using conventional wet impregnation method as reference in the SCR activity test.
2.2 Material characterization
X-ray diffraction (XRD) patterns were recorded on a Rigaku Max-2600 X-ray diffractometer using Cu-Kα radiation. The X-ray tube was operated at 40 kV and 30 mA. The intensity data were collected in a 2θ range of 10–80°. The scanning speed was set at 4° min−1 with a step size of 0.02°. The mean size of the ordered (crystalline) domains (d) was estimated using the Scherrer equation. The equation can be written as , where λ is the X-ray wavelength, β is the line broadening at half the maximum intensity (FWHM) in radians, and θ is the Bragg angle. The morphology and the composition of the samples were acquired by a scanning electron microscope (SEM) and energy dispersive spectrometer (EDS) (Hitachi S4800), respectively. The specific surface areas of the samples were measured by N2 adsorption–desorption isotherms at −196 °C using a Brunauer ASAP-2020 analyzer. Prior to each analysis, the sample was degassed under vacuum at 300 °C for 100 min to remove physically adsorbed impurities.
, where λ is the X-ray wavelength, β is the line broadening at half the maximum intensity (FWHM) in radians, and θ is the Bragg angle. The morphology and the composition of the samples were acquired by a scanning electron microscope (SEM) and energy dispersive spectrometer (EDS) (Hitachi S4800), respectively. The specific surface areas of the samples were measured by N2 adsorption–desorption isotherms at −196 °C using a Brunauer ASAP-2020 analyzer. Prior to each analysis, the sample was degassed under vacuum at 300 °C for 100 min to remove physically adsorbed impurities.
The laser Raman experiments were performed at room temperature. A laser radiating at 532 nm was used as the excitation source, and the laser power of below 0.07 mW was applied at the sample. Before measurements, the samples were well ground and mounted into a spinning holder to avoid thermal damage during the scanning.
X-ray photoelectron spectrometry (XPS) was used to analyze the atomic surface concentration on each catalyst, and the spectra were recorded on a Thermo Fisher Scientific ESCALAB-250 X-ray photoelectron spectrometer using monochromated Al-Kα as a radiation source at 150 W. The powder catalysts were mounted onto the sample holder and degassed overnight at room temperature at a pressure on the order of 10−7 Torr. Binding energies (BE) were measured for C 1s, O 1s, Ti 2p, and Cu 2p. The Auger spectrum for Cu was also recorded. Sample charging effects were eliminated by correcting the observed spectra with the C 1s binding energy (BE) value of 284.6 eV. An estimated error of 0.1 eV can be considered for all the measurements. The O 1s and Cu 2p peaks were deconvoluted using the Gaussian function.
Temperature-programmed desorption of O2 (O2-TPD) was performed using a simultaneous thermal analyzer and the change of gas composition was detected by a quadrupole mass spectrometer. The samples (200 mg, 20–40 mesh) that were placed in the platinum basket were first pretreated in argon for dehydration and removing impurities at 350 °C for 30 min, cooled to 50 °C in argon (30 mL min−1) and saturated with O2/Ar (21 vol% O2 and Ar as the equilibrium gas) at 30 mL min−1 at 50 °C for 60 min. Finally the samples were blown dry in an Ar stream and the desorption was carried out by heating the samples from 50 °C to 1000 °C at a rate of 10 °C min−1 in Ar (30 mL min−1). In the whole process, the desorption curve of oxygen (m/z = 32) and the mass change of the samples were recorded by the QIC-20 quadrupole mass spectrometer and the STA449 thermal analyzer, respectively, and the adsorption and desorption amounts of oxygen were obtained. The argon gas used in test was pre-processed by deep dehydration and deoxygenation treatment and the O2/Ar mixture gas used in the test was pre-processed by deep dehydration treatment.
Temperature-programmed desorption of NO (NO-TPD) was also performed on a fixed-bed reactor, using 200 mg of catalysts sample of 40–60 mesh size. Prior to the TPD experiments, catalysts were pretreated in a continuous flow of Ar gas at 30 mL min−1 flow rate at 350 °C for 30 min, then saturated with the NO/Ar mixture gas (5000 ppm NO, 30 mL min−1) at 50 °C for 30 min. Before the temperature increasing process, the samples were purged with Ar for 1 h at 50 °C. Desorption was carried out by heating the sample in Ar (30 mL min−1) at a heating rate of 10 °C min−1. The released gases during TPD measurements were detected by a quadrupole mass spectrometer. For NO desorption data, the NO (m/z = 30) and NO2 (m/z = 46) signals were detected.
The temperature-programmed reduction of CO (CO-TPR) measurements was performed using the same experimental apparatus as NO-TPD. For the CO-TPR tests, the sample (300 mg) with 40–60 mesh size was placed in the quartz tube. A gas flow (30 mL min−1) containing 10 vol% CO in Ar was used to perform the CO-TPR analysis. Before CO-TPR analysis, the catalyst was pretreated at 350 °C then cooled down to room temperature in Ar, and then conditioned under the reducing gas mixture for 30 min at room temperature before increasing the temperature to 1000 °C at 10 °C min−1. The signal of CO2 (m/z = 44) produced in the CO-TPR process was recorded by a quadrupole mass spectrometer.
Temperature-programmed reduction of H2 (H2-TPR) was carried out using homemade experimental apparatus with a quartz U-tube reactor and the detector used a quadrupole mass spectrometer. The sample (300 mg, 40–60 mesh) was used for each measurement. Before reduction, the sample was pretreated in an Ar stream at 350 °C for 1 h and then cooled to room temperature. The sample was then passed through an H2/Ar mixture (10% H2 by volume), with a linear increase in temperature at a rate of 10 °C min−1. H2 consumption was detected using a mass spectrometer (GAM 200).
In the temperature programmed surface reaction (NO–O2-TPSR) test, the instrument used and method and condition of sample pretreatment was in agreement with that used in NO-TPD test. For the NO–O2-TPSR, the pretreated sample (300 mg, 40–60 mesh) was saturated with the NO/Ar mixture gas (5000 ppm NO, 30 mL min−1) at 50 °C for 30 min and purged with Ar for 1 h. The sample was then passed through O2/Ar mixture (10% O2 by volume) and the NO–O2-TPSR was carried out at a heating rate of 10 °C min−1. The signal of NO2 (m/z = 46) produced in the NO–O2-TPSR was recorded by a quadrupole mass spectrometer.
The in situ diffuse reactance infrared Fourier transform spectroscopy (DRIFTS) experiments were performed on an FTIR spectrometer equipped with a smart collector and an MCT/A detector. Prior to each experiment, the sample was purged in N2 at 500 °C for 1 h.
2.3 Catalytic activity tests
The catalytic activity test for NH3-SCR in the presence of oxygen was carried out at atmospheric pressure in a temperature-programmed reaction system equipped with a fixed bed quartz microreactor with i.d. 8 mm. In total, 0.4 g of catalyst (20–40 mesh) was placed in the reactor in between two glass wool plugs. The feed gas composition was as follows: 600 ppm NO, 600 ppm NH3, 5 vol% O2, 5 vol% H2O (when used), 100 ppm SO2 (when used) and Ar as balance. The total flow rate of the feed gas was 300 mL min−1, corresponding to a gas hourly space velocity (GHSV) of 30000 h−1. The reactor system was heated from room temperature to 400 °C with a heating rate of 4 °C min−1. The NO and NO2 concentrations of the feed gas and products were continually monitored by a chemiluminescence NO/NOx detector. To avoid modest errors caused by the oxidation of ammonia in the converter of the NO/NOx analyzer, an ammonia trap containing phosphoric acid solution was installed before the sample inlet to the chemiluminescence detector. NO conversion was calculated as: 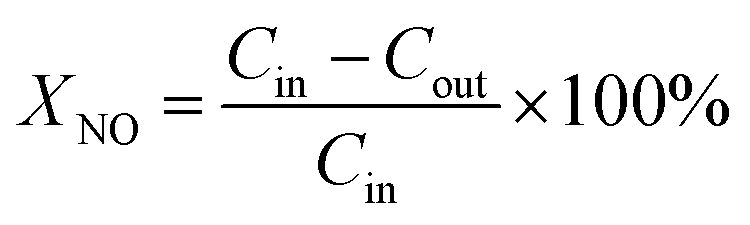 . In the equation, XNO denotes NO conversion and Cin and Cout denotes the inlet and outlet gas concentrations (ppm) of NO, respectively. T85 and T95 indicate the reaction temperatures of NO conversation at 85% and 95%, respectively. The lower the value of T85 and T95 are, the better the activity of the catalysts are.
. In the equation, XNO denotes NO conversion and Cin and Cout denotes the inlet and outlet gas concentrations (ppm) of NO, respectively. T85 and T95 indicate the reaction temperatures of NO conversation at 85% and 95%, respectively. The lower the value of T85 and T95 are, the better the activity of the catalysts are.
3. Results and discussion
3.1 Structure and morphology of TiO2 and Cu(3)/TiO2(T) samples
Fig. 1 shows XRD patterns of pure TiO2(T) and Cu(3)/TiO2(160) in the range of 10–80°. All of diffraction peaks were classified to standard anatase (JCPDS card no. 21-1272) as tetragonal, which is in accordance with previous work.22,24–28 However, when crystallization temperature of TiO2 was higher than 160 °C, another characteristic peak appeared, which belongs to monoclinic TiO2 (JCPDS card no. 46-1238), besides the peaks belonging to tetragonal TiO2. Therefore, when crystallization temperature is higher than 160 °C, TiO2 samples are a mixture of tetragonal and monoclinic TiO2. Owing to the diffraction peaks of tetragonal and monoclinic TiO2 having some overlap, the diffraction peaks of TiO2(160) and TiO2(170) samples become wider in their XRD patterns. TiO2 crystallite size was calculated by Scherrer equation and the results are listed in Table 1. The crystallite size of TiO2 increased with the increasing of crystallization temperature for similar morphology of TiO2. The crystallite size of TiO2(160) and TiO2(170) samples are smaller than TiO2(140) and TiO2(150) owing to the formation of monoclinic TiO2. | ||
| Fig. 1 X-ray powder diffraction patterns of TiO2(T) and Cu(3)/TiO2(160) samples. | ||
| Samples | BET surface area (m2 g−1) | Diameter (nm) | Oxygen adsorption capacity (mg g−1) | ||
|---|---|---|---|---|---|
| Adsorbeda | Superficial | Lattice | |||
| a The number of adsorbed oxygen of catalysts during the stage of isothermal adsorption in O2-TPD test. | |||||
| TiO2(140) | 187.421 | 7.083 | — | — | — |
| TiO2(150) | 182.745 | 8.121 | — | — | — |
| TiO2(160) | 236.778 | 5.936 | — | — | — |
| TiO2(170) | 205.036 | 6.971 | — | — | — |
| Cu(3)/TiO2(140) | 181.618 | 9.963 | 9.67 | 13.73 | 5.81 |
| Cu(3)/TiO2(150) | 178.529 | 11.047 | 12.86 | 15.51 | 5.93 |
| Cu(3)/TiO2(160) | 225.646 | 8.499 | 12.90 | 16.57 | 4.95 |
| Cu(3)/TiO2(170) | 198.131 | 9.218 | 12.81 | 15.55 | 4.58 |
No characteristic diffraction peaks of the copper species was observed in the Cu(3)/TiO2(160) sample and only the characteristic diffraction peaks of TiO2 were identified from the XRD results in Fig. 1. The absence of diffraction peaks from the copper species in Cu(3)/TiO2(160) sample can be explained by the fine dispersion of the species on the surface of TiO2 as well as the low copper content, and/or the amount of Cu species accumulated on TiO2 (3 wt%) is below the XRD detection limit. The same results have been reported previously.27,29 It is noted that compared with TiO2(160) sample, the peak intensity of Cu(3)/TiO2(160) is enhanced owing to fact that Cu(3)/TiO2(160) was calcined again at the same temperature in the preparation process.
Raman spectroscopy was employed to confirm the phases on Cu(3)/TiO2 catalysts. As shown in Fig. 2, all catalysts show typical Raman shifts due to anatase TiO2, which are located at 143, 199, 401, 519, 638 cm−1. The strong peak at 143 cm−1 is attributed to the bending vibration mode of O–Ti–O. The medium peak at 638 cm−1 is assigned to the symmetrical stretching vibration of O–Ti–O. The weak peaks at 401 and 519 cm−1 are corresponding to the symmetric and unsymmetrical bending vibration of O–Ti–O, respectively. The results indicate that the main phase is anatase phase over Cu(3)/TiO2 catalysts. The Raman intensity of the peak at 247 cm−1 increases with elevating the crystallization temperature, which is assigned to the Raman active mode of monoclinic TiO2. It means that monoclinic TiO2 phase become more obvious over Cu(3)/TiO2(160) and Cu(3)/TiO2(170) catalysts, which is consistent with XRD results. It is also noted that the bands attributing to CuO phase in Raman spectra are absent due to its highly dispersion or detection limits.
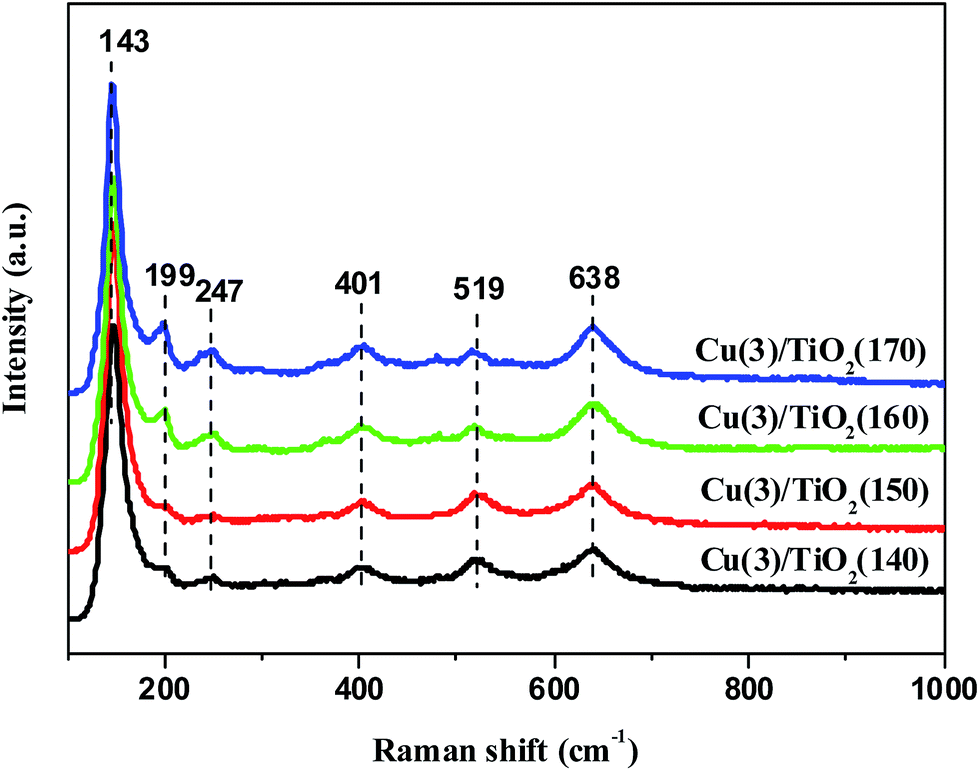 | ||
| Fig. 2 The Raman spectra of Cu(3)/TiO2(T) samples. | ||
Fig. 3 shows SEM results of TiO2(T) (T = 140 °C, 150 °C, 160 °C, 170 °C) and Cu(3)/TiO2(160). The crystallization temperatures remarkably affect on the morphology of TiO2. When they were 140 °C and 150 °C, TiO2 exhibited a columnar structure which assembled by nano-crystals (5–10 nm). At a crystallization temperature of 140 °C, its structure exhibited a little disorder (Fig. 3A); while at 150 °C, the columnar structure exhibited more order (Fig. 3B). When crystallization temperature reached 160 °C, the morphology of TiO2 was substantially changed (Fig. 3C and D). Here, TiO2 is flower-shaped assigned by thin chips in thickness of 5–8 nm. Ye23 and He24 also obtained the flower-shaped TiO2, designated by the nano-thin chips, under the synthetic conditions of 200 °C for 24 hours. Compared with previous work, the preparation methods in this work are easier to perform. Specific surface areas of these samples are approximately 180–237 m2 g−1, which are higher than that achieved from previous work.23–25 The specific surface area of TiO2 synthesized at 160 °C can reach to 236.78 m2 g−1.
 | ||
| Fig. 3 SEM images for TiO2(140) (A), TiO2(150) (B), TiO2(160) (C and D), TiO2(170) (E) and Cu(3)/TiO2(160) (F) and EDS spectrum of the Cu(3)/TiO2(160) (G and H). | ||
The SEM images in Fig. 3F show that the morphology of Cu(3)/TiO2(160) is virtually unchanged after supporting copper species. The EDS result (in Fig. 3G and H) of Cu(3)/TiO2(160) sample indicates they are composed of copper and oxygen-rich phases.
3.2 Surface properties and composition of TiO2 and Cu(3)/TiO2(T) samples
XPS analysis is carried out to elucidate the surface components of Cu(3)/TiO2(T) and TiO2 samples. XPS spectra of Ti 2p, O 1s and Cu 2p of TiO2 and Cu(3)/TiO2(T) sample are shown in Fig. 4. As shows in Fig. 4A, TiO2 and Cu(3)/TiO2(T) samples exhibit two binding energy peaks of the Ti 2p3/2 and Ti 2p1/2 at approximately 458.10 and 463.9 eV, respectively, which are the characteristics peaks of Ti4+ species.3,18,30 Compared with pure TiO2 carriers, the central peaks of Cu(3)/TiO2(T) catalysts are similar. However, the broadening in the binding energy of the Ti 2p on Cu(3)/TiO2(T) may arise from the strong interaction between the copper species and TiO2 support.3,17,29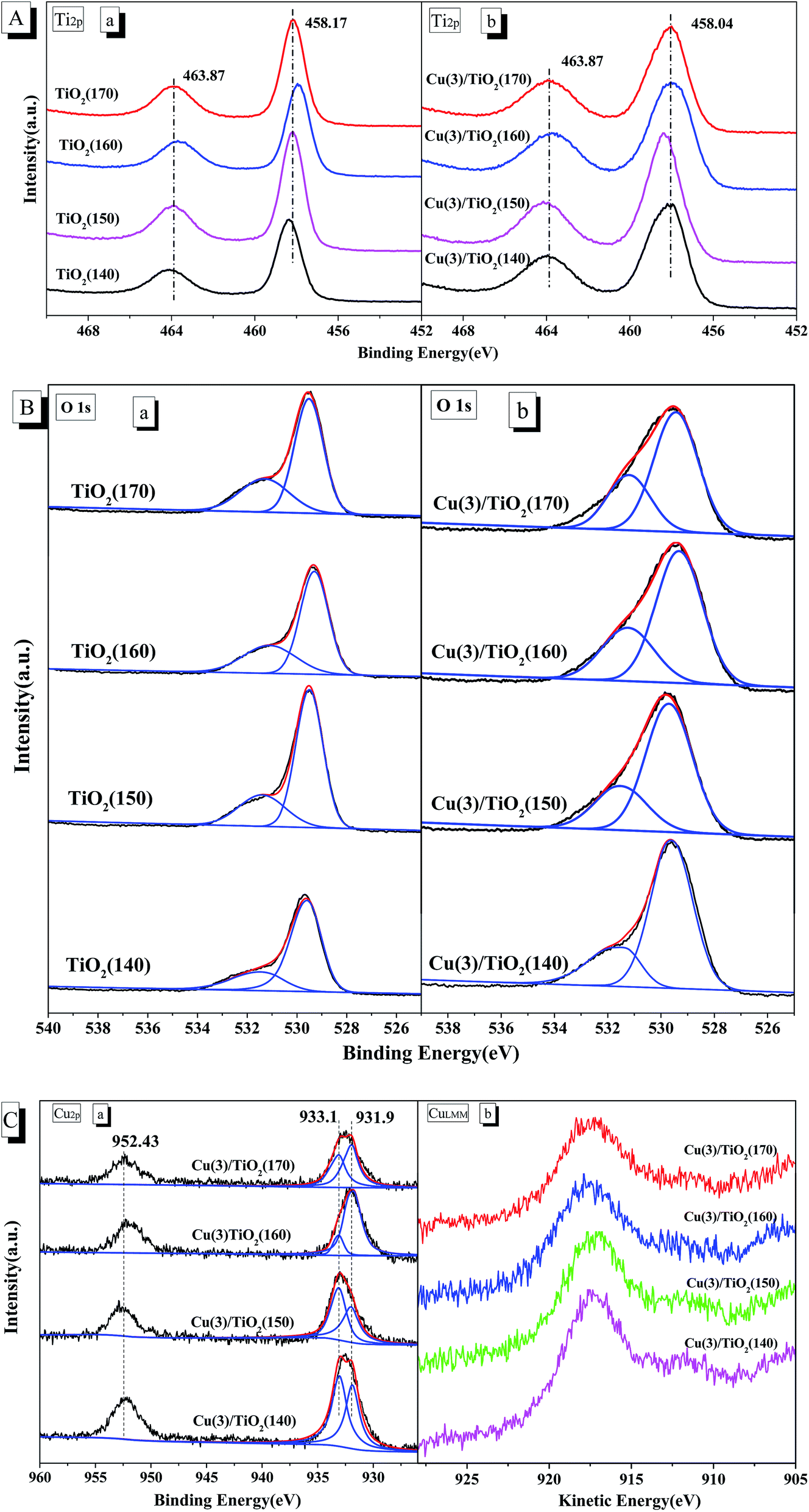 | ||
| Fig. 4 The XPS spectra of Ti 2p (A), O 1s (B), Cu 2p (C(a)) and Cu LMM (C(b)) spectra of TiO2(T) and Cu(3)/TiO2(T) samples. | ||
The broad and overlapping peaks (545–521 eV) of O 1s on TiO2 and Cu(3)/TiO2(T) samples are revealed in Fig. 4B. The O 1s peaks were deconvoluted into two peaks at approximately 529.5 and 531.3 eV. The peaks at the low binding energy (529.5 eV) can be ascribed to the lattice oxygen O2− (marked as Oβ) and the peak at the higher binding energy (531.3 eV) can be attributed to the adsorbed oxygen (designated as Oα), such as O2− or O−, −OH groups, and oxygen vacancies.4,6,16,28 As shown in Fig. 4B, the O 1s profile of Cu(3)/TiO2(T) sample is broader than TiO2 because of the overlapping contribution of the oxygen species from TiO2 and promoted copper oxides. According to the peak area, the ratios of adsorbed oxygen (Oα) to total oxygen (Oα + Oβ) were calculated and the results are listed in Table 2. The O 1s peak positions show that the amount and type of surface oxygen species on TiO2 were affected by the crystallization temperature. The percentage of adsorbed oxygen (Oα) to total oxygen (Oα + Oβ) of the Cu(3)/TiO2(T) and TiO2 samples first increases with the increasing of the crystallization temperature and reached a maximum when the crystallization temperature was 160 °C and the percentage is 35.48% and 35.23%, respectively, which indeed results in more surface oxygen vacancies. The surface oxygen species are the main oxidative intermediate and are highly active in oxidation reactions owing to the higher mobility than lattice oxygen species.16,28 Consequently, the higher Oα/(Oα + Oβ) ratio on Cu(3)/TiO2(T) is favorable for NO oxidation to NO2 in the SCR reaction.6,16
| Samples | Surface element composition (at%) | Surface atomic composition | Auger parameter (eV) | |||
|---|---|---|---|---|---|---|
| O | Cu | Ti | Oα/(Oα + Oβ) (%) | Cu+/Cu2+ (Cu2p) | ||
| TiO2(140) | 50.71 | — | 16.32 | 30.53 | — | — |
| TiO2(150) | 56.47 | — | 18.59 | 33.87 | — | — |
| TiO2(160) | 54.40 | — | 17.58 | 35.48 | — | — |
| TiO2(170) | 52.30 | — | 17.60 | 35.34 | — | — |
| Cu(3)/TiO2(140) | 34.15 | 1.72 | 8.51 | 25.75 | 1.32 | 1849.3 |
| Cu(3)/TiO2(150) | 44.80 | 1.48 | 15.59 | 33.30 | 1.54 | 1849.2 |
| Cu(3)/TiO2(160) | 51.19 | 1.87 | 14.84 | 35.23 | 3.56 | 1849.2 |
| Cu(3)/TiO2(170) | 47.50 | 1.38 | 17.50 | 34.98 | 1.73 | 1849.2 |
Cu 2p spectra and XPS spectra of Cu LMM of Cu(3)/TiO2(T) samples are displayed in Fig. 4C. The binding energy in the range of 933.1–934.5 eV and the characteristic shake-up peaks at a binding energy of 944 eV can be attributed to Cu2+, and the binding energy in the range of 928.9–932.5 eV and the absence of shake-up peaks can be assigned to Cu+ or Cu0.2,30–35 Cu 2p spectrum of Cu(3)/TiO2(T) samples (Fig. 4C(a)) shows two main peaks attributed to Cu 2p3/2 and Cu 2p1/2 at approximately 932.5 and 952.4 eV, respectively. For Cu(3)/TiO2(T) samples, Cu 2p3/2 peaks show considerable asymmetry. Cu 2p3/2 peaks can be fitted to two peaks, which represent BEs at 933.1 and 931.9 eV corresponding to the Cu2+ and Cu+/Cu0 species, respectively. The absence of the shake-up peak centered at 944 eV for the Cu2+ species may be as a result of the low copper loading and the less Cu2+ species and the highly dispersed amorphous CuO.33 As Cu 2p3/2 binding energies of Cu+ and Cu0 are almost identical, it is difficult to distinguish between Cu+ and Cu0 from the XPS spectra, and one can only distinguish between these two valence states by Auger analysis.31,35 Fig. 4C(b) shows an Auger kinetic energy peak, ranging from 914.4 to 924.4 eV, is observed in the XPS data for Cu(3)/TiO2(T) samples. The Auger parameter, which is calculated as the sum of the Cu 2p binding energy and Auger peak kinetic energy, is included in Table 2. The values of the Auger parameter of most of the catalysts are approximately 1849.2 eV, and these values confirm the presence of Cu+ cations, which further confirm that there was no Cu0.32 The result of Cu 2p spectra and Cu LMM (Fig. 4C) illustrate the presence of the major Cu2+ and Cu+ species. Thus, it could be inferred that CuO and Cu2O species are in Cu(3)/TiO2(T) catalysts, where the copper species in Cu(3)/TiO2(T) sample exist as Cu+ and Cu2+.2,16,29,34 Kannekanti et al.27 observed that copper species exist as Cu+ when Cu(3)/TiO2(T) sample was calcined at higher temperature (450 °C), and as Cu2+ at lower temperature (350 °C). The existence of both Cu2+ and Cu+ species is favorable for the redox cycle reaction of the SCR of NOx with NH3.16 Ndong et al.28 proposed that Cu2O should be more negative than CuO, and Cu2O/TiO2(T) photocatalysts exhibit a higher photocatalytic activity compared with CuO/TiO2(T). The relative ratios of copper species are calculated by the area ratio of corresponding characteristic peaks of Cu 2p3/2, and are listed in Table 2. XPS results indicate that the surface atomic ratio of Cu+/Cu2+ increases monotonically with the increasing of the crystallization temperature and reaches a maximum (Cu+/Cu2+ = 3.56) at 160 °C. More Cu+ cations in Cu(3)/TiO2(160) may exhibit a higher catalytic activity compared with other samples owing to them being more negative.
3.3 NO-TPD and O2-TPD analysis of the Cu(3)/TiO2(T) samples
After NO was adsorbed on the surface of Cu(3)/TiO2(T) thermal desorption proceeded as a dissociation reaction, and NO desorption species were detected by mass spectrometry (NO, m/z = 30) (Fig. 5A). NO desorption of Cu(3)/TiO2(T) exhibited four desorption peaks, that is, four NO adsorption centers, and the desorption peak temperatures were at approximately 95, 118, 135 and 350 °C.36 The first one was attributed to NO desorption on weak sites, the second and third ones were assigned to desorption on the strong sites, and the peak at higher temperature can be assigned to the stronger sites. The amount of NO desorption for Cu(3)/TiO2(140) is very small and the dissociation activity of NO is low. The amount of desorption of NO for the other three samples is considerable, but the desorption peak temperatures of Cu(3)/TiO2(150) and Cu(3)/TiO2(160) are lower than that of Cu(3)/TiO2(170). The former two exhibit a better dissociation activity than the latter. It is worth mentioning that the desorption peak of NO around 350 °C of Cu(3)/TiO2(160) disappears or moves to a lower temperature. It is worth noting that NO2 signals are detected in the NO temperature-programmed desorption process, which shows that NO has been oxidized to NO2 (Fig. 5B). The four desorption peaks of NO2 are detected for all samples, which shows that the different NO oxidation centers exist on the catalysts. According to the position and peak area of NO2 peaks, it is clear that NO is more easily oxidized into NO2 on Cu(3)/TiO2(160) sample. The generation of NO2 is crucial for the fast SCR happened on the catalyst surface. The phenomenon of NO oxidized into NO2 is confirmed in the NO–O2-TPSR test (Fig. 5C) on Cu(3)/TiO2(160) sample. All these results show that Cu(3)/TiO2(160) sample exhibits the best dissociation activity, which is in agreement with the higher activity of the catalyst.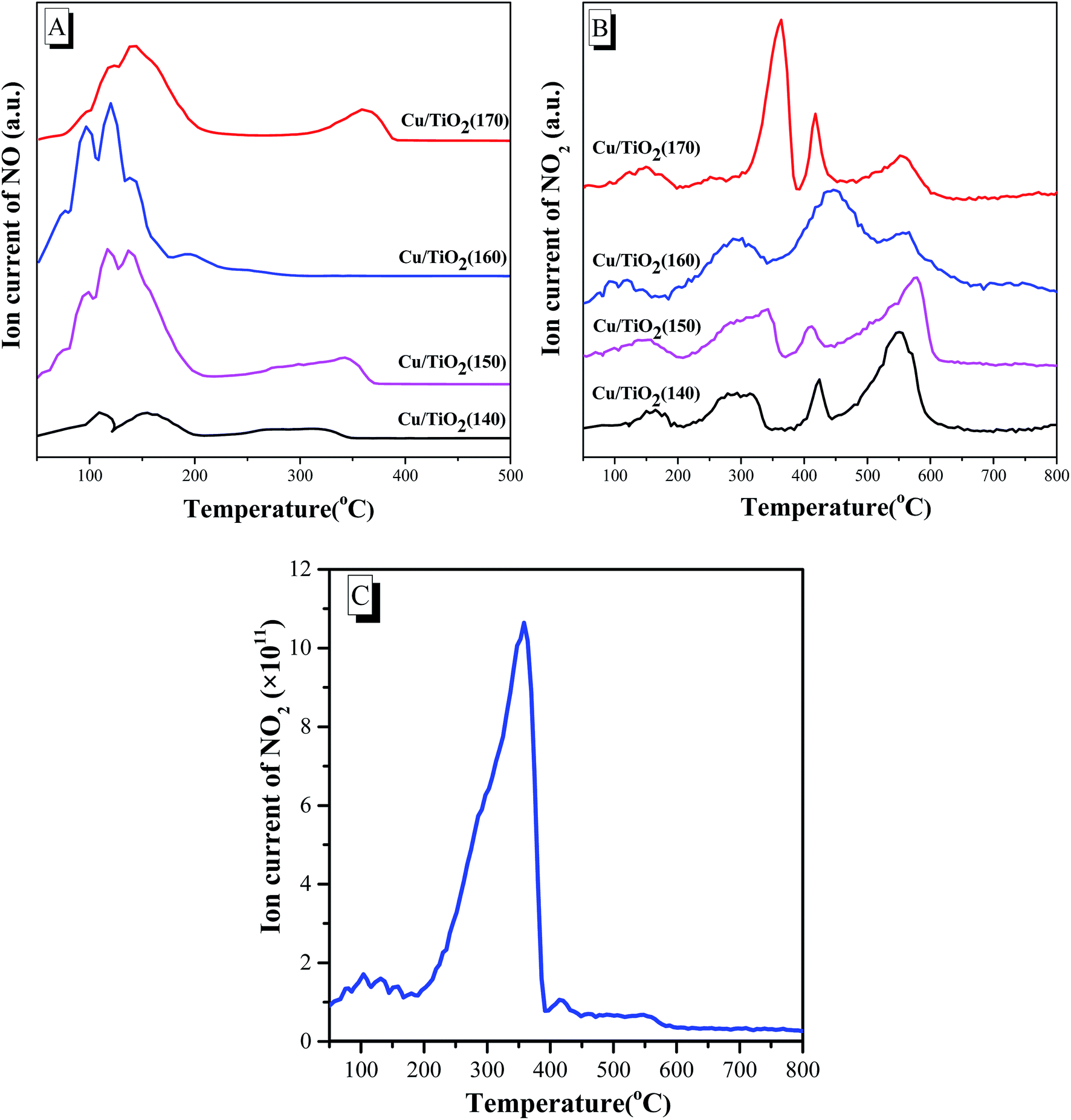 | ||
| Fig. 5 The NO-TPD patterns (A and B) of Cu(3)/TiO2(T) samples and the NO–O2-TPSR patterns (C) of the Cu(3)/TiO2(160) sample. | ||
O2-TPD experiments were carried out to gain insight into the nature of the surface oxygen species involved, and the results are shown in Fig. 6. When the temperature is lower than 800 °C, three very weak and broad peaks appear, as shown in Fig. 6A. Approximately 1.3–1.5% of the continuous weight loss was detected below 800 °C, as shown in Fig. 6B. The results may arise from the presence of different adsorption strengths of the same oxygen species. According to the literature,37,38 the desorption peaks at lower temperatures are generally ascribed to superficial oxygen species and are related to molecular O2, O2− and O22 adsorbed on oxygen vacancies. The lattice oxygen desorption temperature is usually above 800 °C37,38 and the significant desorption peak of lattice oxygen and the weight loss of the samples is detected. It is noted that the lattice oxygen desorption peak is composed of two overlapping peaks. It indicates that there may be two kinds of properties of the lattice oxygen. Therefore, two kinds of copper oxide species show that the copper species exist in Cu2O and CuO. This is consistent with the results from XPS analysis.
 | ||
| Fig. 6 The O2-TPD patterns of the Cu(3)/TiO2(T) samples. | ||
According to the change of sample mass, the amounts of adsorbed oxygen, surface adsorbed oxygen and lattice oxygen, are obtained, and the results are listed in Table 1. From Table 1, the amount of oxygen in the samples is related to the crystallization temperature in the preparation process of TiO2. As the crystallization temperature of TiO2 is increased, the amount of adsorption and surface oxygen on the Cu(3)/TiO2(T) samples increases monotonously. The oxygen quantity reaches the maximum when the crystallization temperature is 160 °C, which is related to the oxygen vacancy of the samples. With the increasing of the crystallization temperature, the amount of lattice oxygen in Cu(3)/TiO2(T) samples reduces and the desorption temperature of Cu(3)/TiO2(T) samples is an inverted parabola (see Fig. 6B), so the desorption temperature of Cu(3)/TiO2(150) sample exhibits a minimum value, and Cu(3)/TiO2(160) sample is the second. The results of O2-TPD show that there are more oxygen vacancies and a better activity of the lattice oxygen in Cu(3)/TiO2(160) sample.
3.4 H2-TPR and CO-TPR and analysis of the Cu(3)/TiO2(T) samples
Temperature-programmed reduction (TPR) is a powerful tool to investigate the reducibility and identify the different surface oxygen species. The reducibility of copper species in Cu(3)/TiO2(T) catalysts was investigated by H2-TPR and CO-TPR experiments.H2-TPR results reported previously on a TiO2 supported copper oxide3,31,36,39 showed that three or four reduction peaks (α, β, γ and δ) could be detected. The α peak results from the highly dispersed copper species, the β peak is originated from the fine grain copper species (i.e., short-range order but not crystallites), the γ peak is come from the copper species crystallites, and the δ peak results from the interactions between copper oxide and TiO2. When the concentration of the copper species is lower or highly dispersed, the γ and δ peaks are not detected.33,36 H2-TPR results are shown in Fig. 7. Two reduction peaks (α and β) between 150–300 °C occurred in the profiles for H2-TPR of Cu(3)/TiO2(T) catalysts, and the γ and δ peaks were not detected (Fig. 7). The results indicated that the copper species are highly dispersed on TiO2.36,39 In comparison with the α peaks (high dispersion copper species), the β peak (fine grain copper species) is relatively weak, which indicates that most of the copper species exist in the form of the highly dispersed species on the TiO2. This is consistent with XRD, XPS and BET results. The large specific surface area aids in the copper species dispersion on TiO2. Therefore, the improvement in catalytic activities is proposed to be related to the high dispersion of the copper species.36 The reducibility of Cu(3)/TiO2(T) catalysts was investigated by employing also CO as reducing agent in TPR tests, and results are shown in Fig. 8. All samples exhibit three partly overlapping peaks of CO2 release in the temperature range of 50–700 °C in the CO-TPR process, and similar results have been observed in the literature.34,40 Several processes lead to CO2 production, such as CO + Os → CO2 reduction process (where Os denotes a surface oxygen atom), CO and surface hydroxyl groups and CO disproportionation (Boudouard reaction: 2CO → CO2 + C, if the reaction takes place, the sample color will change).40,41 There is no Boudouard reaction because the sample color does not change and the baseline returned to the initial position during CO-TPR process.34 The peaks (α) below 300 °C can be attributed to the reaction between CO and Os or surface hydroxyl groups. The peaks (β) at approximately 400 °C are attributed to the reduction of CuO to Cu0 with CO occurs in one single step,34 and the peaks(γ) about 600 °C are the reduction of Cu2O to Cu0. According to Fig. 7, the ratio of β to γ peak area in CO-TPR over Cu(3)/TiO2(T) increases with the rise of the crystallization temperature until it reached to 160 °C, where the a ratio of β to γ peak area is the greatest. The results show there are more Cu2O species in Cu(3)/TiO2(160) sample, which is highly consistent with XPS results.
 | ||
| Fig. 7 The H2-TPR patterns of the Cu(3)/TiO2(T) samples. | ||
 | ||
| Fig. 8 The CO-TPR patterns of Cu(3)/TiO2(T) samples. | ||
3.5 Catalytic activity tests
The catalytic performances of Cu(3)/TiO2(T) catalysts are shown in Fig. 9A. NO conversion for Cu(3)/TiO2(T) catalysts is exhibited in the temperature interval from 100 to 400 °C. For all catalysts, NO conversion increases when the temperature increases until the conversion approaches 100% and reduces at higher temperature. Xie12 suggested that the decline of SCR activity at high temperature should be caused by two reasons: first, part of inlet NO can only react with insufficient adsorbed NH3 species, which is partially consumed in NH3 oxidation. Second, NO is produced during NH3 oxidation reaction, directly decreasing NO conversion. For all catalysts, NO conversion is different at the same Cu-loading, which indicates that NH3-SCR activity is limited by the properties of TiO2 carrier, that is, the differences of TiO2 in morphology and structure. As the crystallization temperature in carrier TiO2 preparation is increased, NH3-SCR activity of Cu(3)/TiO2(T) catalysts monotonically increases. The activity of Cu(3)/TiO2(160) catalyst is best, T85 and T95 is 160 °C and 170 °C, respectively, and the temperature windows of NO conversion over 95% is from approximately 170 °C to 310 °C. Compared with conventional V2O5–WO3/TiO2 catalyst, the low temperature activity of Cu(3)/TiO2(160) catalyst is higher. Fig. 9D shows the NO conversion as a function of reaction temperatures in the presence of 5% H2O or 100 ppm SO2 for Cu(3)/TiO2(160) catalysts. It can be seen that NO conversion over Cu(3)/TiO2(160) catalyst slightly decreased in the presence of 5% H2O in the whole temperature window. The decreased activity may be caused by the competitive adsorption between H2O and NH3 on the acid sites. SO2 shows the complicated effects on the activity of Cu(3)/TiO2(160) catalyst. It inhibited the catalytic activity seriously below 300 °C, while it improved the catalytic activity at high temperature. Compared with V–W/Ti catalyst, this catalyst system may be not suitable for low temperature applications due to its low activity in the presence of SO2. The presence of SO2 in the feed gas induced a significant decrease in NOx conversion over Cu(3)/TiO2(160) catalyst at low temperature, which might be related to the deposition of highly thermally stable ammonium sulfate on the surface of the catalyst, blocking the active sites. The catalytic activity results indicate that further modifications are still urgently needed with an aim to enhance the poisoning resistance of the catalysts. | ||
| Fig. 9 The NH3-SCR activity for NO conversions as a function of reaction temperature for Cu/TiO2. Conditions: 600 ppm NO, 600 ppm NH3, 5 vol% O2, 5% H2O (when used), 100 ppm SO2 (when used), and Ar to balance; flow rate: 300 cm3 min−1; weight of catalyst: 0.4 g. | ||
On TiO2(160) nanospheres carrier, the effects of copper loading amount and different metal active components on the activity of SCR catalyst were investigated (Fig. 9B). The results showed that with the increasing of copper contents, the catalyst activity first increases and reached an optimal activity when the copper loading amount was 3 wt%. It indicates that Cu(3)/TiO2(160) sample gives the good catalytic activity. The SCR catalytic activities of Cu(3)/TiO2(160) and other metal oxides doped TiO2(160) systems are compared (Fig. 9C). The catalysts of metal oxide of iron, cobalt or cerium doped TiO2(160) are used for NH3-SCR reaction. The different metal oxide species in the catalyst remarkably affect the catalytic activity.
3.6 Reaction mechanism
 | ||
| Fig. 10 In situ DRIFTS of NH3 desorption on Cu(3)/TiO2(140) (A) and Cu(3)/TiO2(160) (B) measured at 30–300 °C. | ||
Similarly with regard to Cu(3)/TiO2(160) (Fig. 10B), the asymmetric and symmetric bending vibrations of the N–H bond in NH3 coordinately linked to a Lewis acid site can be observed at 1599, 1226 and 1174 cm−1. Compared with Cu(3)/TiO2(140), the band for symmetric bending vibrations N–H bond in NH3 coordinately linked to a Lewis acid site at 1174 cm−1 can still be observed even at 300 °C. This indicates that the ammonia adsorbed on Lewis acid sites over Cu(3)/TiO2(160) is much stronger than that on Cu(3)/TiO2(140), which is beneficial to the activation of NH3 and the selective catalytic reduction of NO by NH3.
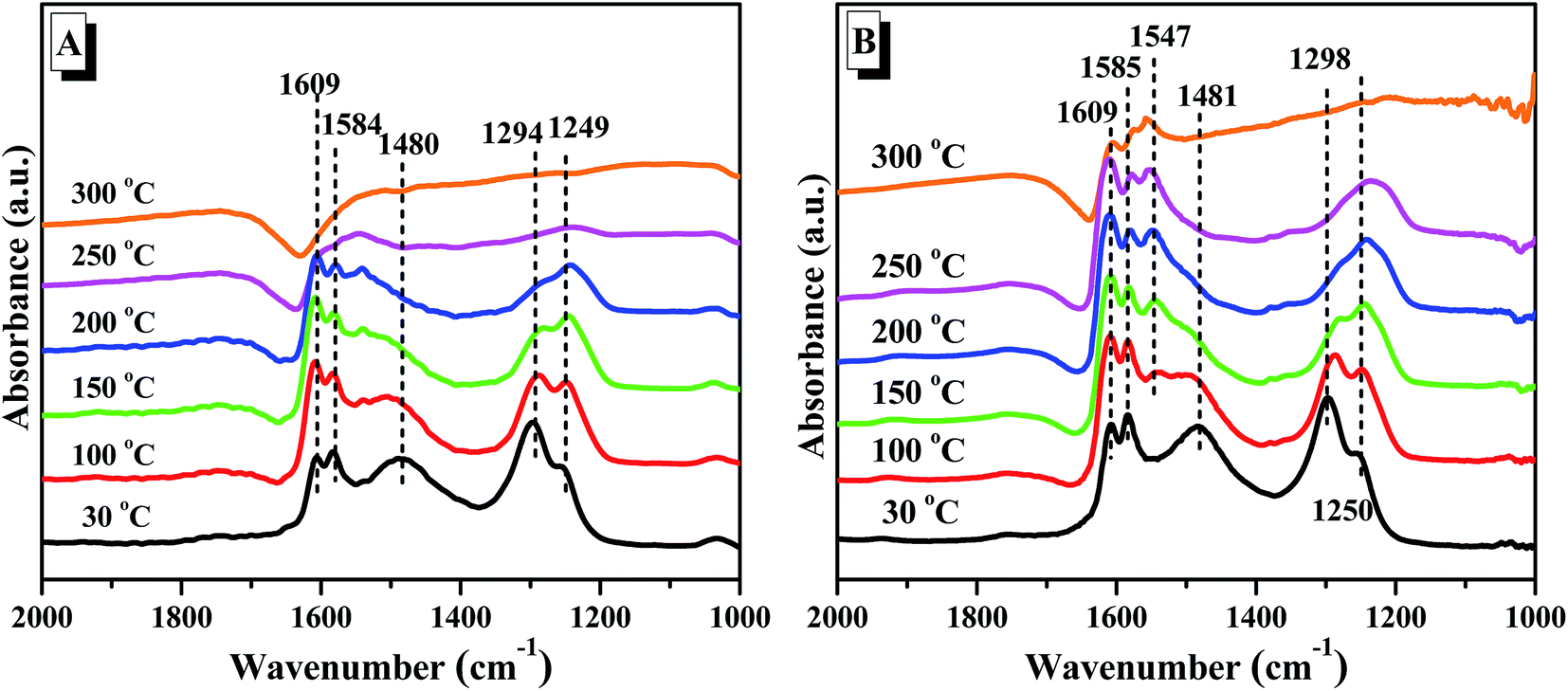 | ||
| Fig. 11 In situ DRIFTS of NO + O2 desorption on Cu(3)/TiO2(140) (A) and Cu(3)/TiO2(160) (B) measured at 30–300 °C. | ||
 | ||
| Fig. 12 In situ DRIFTS over Cu(3)/TiO2(140) (A) and Cu(3)/TiO2(160) (B) as a function of time in a flow of NO + O2 after the catalysts were pre-exposed to a flow of NH3 for 60 min followed by N2 purging for 30 min at 150 °C. | ||
 | ||
| Fig. 13 In situ DRIFTS over Cu(3)/TiO2(140) (A) and Cu(3)/TiO2(160) (B) as a function of time in a flow of NH3 after the catalysts were pre-exposed to a flow of NO + O2 for 60 min followed by N2 purging for 30 min at 150 °C. | ||
As for the reaction mechanism, including the “standard SCR” reaction (eqn (1)), “fast SCR” reaction (eqn (2)) and “slow SCR” or “NO2-SCR” reaction (eqn (2)), consensus has been reached. Usually, the reactants must first be adsorbed onto the surface of the catalyst, and then activated for the reaction to proceed.49,50 Therefore, the reaction must be carried out in two steps, that is, adsorption and activation. The Eley–Rideal (E–R) and Langmuir–Hinshelwood (L–H) mechanisms are applicable for the NH3-SCR reaction. In the E–R mechanism, NO and NO2 in the gas or weakly adsorbed react with ammonia adsorbed on the acidic center of the catalyst surface and is activated to form N2 and H2O. In this process, the active oxygen on catalyst surface is consumed and the oxygen in the gas phase is passed through the catalyst to renew the surface oxygen, so as to complete the catalytic cycle. In L–H mechanism, NH3 and NO are adsorbed on the adjacent active site of the catalyst surface, and the active site is reduced by NH3 at the same time. Adsorbed ammonia is activated to form M–NH2 and/or M–NH3,7,49,50 and NO is activated as M–NO. M–NH3 species can react with M–NO to form N2 and H2O, and M–NH2 reacts with M–NO and lattice oxygen to form N2O. Finally, the reduced active site is re-oxidized by O2, and the catalytic cycle is complete. This shows that these features of the catalysts, such as the number of actives sites, redox properties and adsorption capacity for NH3 and NO, are very important for the NH3-SCR reaction. SEM results show that the flower-like TiO2 synthesized at 160 °C has the biggest specific surface area, so copper species could be highly dispersed on TiO2 surface and more active sites can be exposed, which leads to an enhancement of the adsorption capacity. This is confirmed by the results of NO-TPD and O2-TPD, and XPS. It is reported that the presence of Cu+ species is beneficial to the adsorption and activation of NO.51–53 This is consistent with XPS and activity test results. Thus, in situ DRIFTS and other characteristic results revealed that the Lewis acid sites are involved in NH3-SCR reaction and NO2 species is a principal intermediate following NO adsorption on Cu(3)/TiO2(T) catalysts, which may facilitate the “fast” SCR reaction at low temperature.
4. Conclusions
Nanosphere TiO2 was synthesized by the solvothermal method at the different crystallization temperatures (140 °C, 150 °C, 160 °C and 170 °C) and these TiO2 samples were used as the carriers to prepare Cu/TiO2(T) catalysts with a copper loading of 3 wt% for selective catalytic reduction of NO with NH3 at low temperature. The structure and morphology were affected by the crystallization temperature. When the crystallization temperature is 160 °C, the largest specific surface area at 236.78 m2 g−1 is obtained.The differences of TiO2 in the structure and morphology affect the catalytic performances of Cu(3)/TiO2(T) catalysts for NH3-SCR reaction. Copper species in Cu(3)/TiO2(T) samples exist as Cu+ and Cu2+, and the greater number of Cu+ cations in TiO2(160) carrier is beneficial to the adsorption of NO. Cu(3)/TiO2(160) catalyst shows better redox and NO adsorption abilities. Thus, it presents a good activity for the selective catalytic reduction of NO with NH3 at low temperature.
In situ DRIFTS results revealed that Lewis acid sites are involved in NH3-SCR reaction and NO2 species is a principal intermediate following NO adsorption on Cu/TiO2 catalysts, which may facilitate the “fast” SCR reaction at low temperature.
Conflict of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21673290, 21343009, 21376261), the State Key Laboratory of Heavy Oil Processing (SKLHOP201501) and the Funding Project for Academic Human Resources Development in Institutions of Higher Learning under the Jurisdiction of Beijing Municipality (PHR20110517).References
- D. Wang, L. Zhang, K. Kamasamudram and W. S. Epling, ACS Catal., 2013, 3, 871–881 CrossRef CAS.
- Z. C. Si, D. Weng, X. D. Wu, J. Li and G. Li, J. Catal., 2010, 271, 43–51 CrossRef CAS.
- X. J. Yao, L. Zhang, L. L. Li, L. C. Liu, Y. Cao, X. Dong, F. Gao, Y. Deng, C. J. Tang, Z. Chen, L. Dong and Y. Chen, Appl. Catal., B, 2014, 150, 315–329 CrossRef.
- Z. C. Si, D. Weng, X. D. Wu, Y. Jiang and B. Wang, Catal. Sci. Technol., 2011, 1, 453–461 CAS.
- B. Li, Z. Ren, Y. Z. X. Ma, X. D. Huang, F. Liu, X. B. Zhang and H. S. Yang, Catal. Sci. Technol., 2016, 6, 1719–1725 CAS.
- L. Čapek, J. Dědeček, B. Wichterlová, L. Cider, E. Jobson and V. Tokarová, Appl. Catal., B, 2005, 60, 147–153 CrossRef.
- F. Bin, C. L. Song, G. Lv, J. O. Song, S. H. Wu and X. D. Li, Appl. Catal., B, 2014, 150–151, 532–543 CrossRef CAS.
- V. F. Kispersky, A. J. Kropf, F. H. Ribeiro and J. T. Miller, Phys. Chem. Chem. Phys., 2012, 14, 2229–2238 RSC.
- L. Lisi, R. Pironea, G. Russoc, N. Santamariac and V. Stanzione, Appl. Catal., A, 2012, 413–414, 117–131 CrossRef CAS.
- P. N. Panahi, A. Niaei, D. Salari, S. M. Mousavi and G. Delahay, J. Environ. Sci., 2015, 35, 135–143 CrossRef PubMed.
- J. Szanyi, J. H. Kwak, H. Y. Zhu and C. H. F. Peden, Phys. Chem. Chem. Phys., 2013, 15, 2368–2380 RSC.
- L. J. Xie, F. D. Liu, L. M. Ren, X. Y. Shi, F. S. Xiao and H. He, Environ. Sci. Technol., 2014, 48, 566–572 CrossRef CAS PubMed.
- J. C. Wang, Z. L. Peng, H. Qiao, L. Han, W. R. Bao, L. P. Chang, G. Feng and W. Liu, RSC Adv., 2014, 4, 42403–42411 RSC.
- R. R. Zhang, Y. H. Li and T. L. Zhen, RSC Adv., 2014, 4, 52130–52139 RSC.
- R. Baran, F. Averseng, D. Wierzbicki, K. Chalupka, J. M. Krafft, T. Grzybek and S. Dzwigaj, Appl. Catal., A, 2016, 523, 332–342 CrossRef CAS.
- T. Boningari, D. K. Pappas, P. R. Ettireddy, A. Kotrba and P. G. Smirniotis, Ind. Eng. Chem. Res., 2015, 54, 2261–2273 CrossRef CAS.
- P. M. Sreekanth, D. A. P. And and P. G. Smirniotis, Ind. Eng. Chem. Res., 2006, 45, 6444–6449 CrossRef CAS.
- P. Sangpour, F. Hashemi and A. Z. Moshfegh, J. Phys. Chem. C, 2010, 114, 13955–13961 CAS.
- S. J. Liu, Z. Y. Liu, Z. P. Zhu and H. X. Niu, J. Fuel Chem. Technol., 1999, 27, 192–198 Search PubMed.
- H. Teng, L. Y. Hsu, Y. C. Lai, T. Hsisheng, H. Li-Yeh and C. L. Yu, Environ. Sci. Technol., 2001, 35, 2369–2374 CrossRef CAS PubMed.
- K. Zhuang, J. Qiu, F. S. Tang, B. L. Xu and Y. N. Fan, Phys. Chem. Chem. Phys., 2011, 13, 4463–4469 RSC.
- L. Gao, X. R. Li, H. Hu, G. J. Li, H. W. Liu and Y. Yu, Electrochim. Acta, 2014, 120, 231–239 CrossRef CAS.
- J. F. Ye, W. Liu, J. G. Cai, S. A. Chen, X. W. Zhao, H. H. Zhou and L. M. Qi, J. Am. Chem. Soc., 2011, 133, 933–940 CrossRef CAS PubMed.
- F. He, J. L. Li, T. Li and G. X. Li, Chem. Eng. J., 2014, 237, 312–321 CrossRef CAS.
- S. F. Chen, H. Q. Wang, L. J. Zhu, J. F. Li and J. F. Sun, Appl. Surf. Sci., 2014, 321, 86–93 CrossRef CAS.
- C. X. Wang, L. W. Yin, L. Y. Zhang, Y. X. Qi, N. Lun and N. N. Liu, Langmuir, 2010, 26, 12841–12848 CrossRef CAS PubMed.
- K. Lalitha, G. Sadanandam, V. D. Kumari, M. Subrahmanyam, B. Sreedhar and N. Y. Hebalkar, J. Phys. Chem. C, 2010, 114, 22181–22189 CAS.
- L. B. B. Ndong, M. P. Ibondou, X. G. Gu, S. G. Lu, Z. F. Qiu, Q. Sui and S. M. Mbadinga, Ind. Eng. Chem. Res., 2014, 53, 1368–1376 CrossRef.
- B. F. Xin, P. Wang, D. D. Ding, J. Liu, Z. Y. Ren and H. G. Fu, Appl. Surf. Sci., 2008, 254, 2569–2574 CrossRef CAS.
- M. S. Kim, S. H. Chung, C. J. Yoo, M. S. Lee, I. H. Cho, D. W. Lee and K. Y. Lee, Appl. Catal., B, 2013, 142–143, 354–361 CrossRef CAS.
- G. Q. Zhang, Z. Li, H. Y. Zheng, T. J. Fu, Y. B. Ju and Y. C. Wang, Appl. Catal., B, 2015, 179, 95–105 CrossRef CAS.
- B. Pereda-Ayo, U. De La Torre, M. J. Illan-Gomez, A. Bueno-Lopez and J. R. Gonzalez-Velasco, Appl. Catal., B, 2014, 147, 420–428 CrossRef CAS.
- K. V. R. Chary, S. Guggilla Vidya, N. Dhachapally, S. Kottapalli Kalyana and S. Bojja, J. Phys. Chem. B, 2005, 109, 9437–9444 CrossRef CAS PubMed.
- H. B. Zou, S. Z. Chen and W. M. Lin, J. Nat. Gas Chem., 2008, 17, 208–211 CrossRef CAS.
- X. L. Zheng, H. Q. Lin, J. W. Zheng, X. P. Duan and Y. Z. Yuan, ACS Catal., 2013, 3, 2738–2749 CrossRef CAS.
- X. Y. Jiang, G. H. Ding, L. P. Lou, Y. G. Chen and X. M. Zheng, J. Mol. Catal. A: Chem., 2004, 218, 187–195 CrossRef CAS.
- L. H. Dong, Y. X. Tang, B. Li, L. Y. Zhou, F. Z. Gong, H. X. He, B. Z. Sun, C. J. Tang, F. Gao and L. Dong, Appl. Catal., B, 2016, 180, 451–462 CrossRef CAS.
- Q. Liang, X. D. Wu, D. Weng and H. B. Xu, Catal. Today, 2008, 139, 113–118 CrossRef CAS.
- Z. X. Ma, H. S. Yang, F. Liu and X. B. Zhang, Appl. Catal., A, 2013, 467, 450–455 CrossRef CAS.
- C. A. Sierra-Pereira and E. A. Urquieta-Gonzalez, Fuel, 2014, 118, 137–147 CrossRef CAS.
- A. Martínezarias, R. Cataluña, J. C. Conesa and J. Soria, J. Phys. Chem. B, 1998, 102, 809–817 CrossRef.
- A. Zecchina, L. Marchese, S. Bordiga, C. Paze and E. Gianotti, J. Phys. Chem. B, 1997, 101, 10128–10135 CrossRef CAS.
- L. Ma, Y. Cheng, G. Cavataio, R. W. McCabe, L. Fu and J. H. Li, Appl. Catal., B, 2014, 156, 428–437 CrossRef.
- F. S. Tang, B. L. Xu, H. H. Shi, J. H. Qiu and Y. N. Fan, Appl. Catal., B, 2010, 94, 71–76 CrossRef CAS.
- K. Cheng, W. Y. Song, Y. Cheng, J. Liu, Z. Zhao and Y. C. Wei, Catal. Sci. Technol., 2016, 6, 4478–4490 CAS.
- Q. Li, X. X. Hou, H. S. Yang, Z. X. Ma, J. W. Zheng, F. Liu, X. B. Zhang and Z. Y. Yuan, J. Mol. Catal. A: Chem., 2012, 356, 121–127 CrossRef CAS.
- C. Sedlmair, K. Seshan, A. Jentys and J. A. Lercher, J. Catal., 2003, 214, 308–316 CrossRef CAS.
- J. J. Yu, Z. Jiang, L. Zhu, Z. P. Hao and Z. P. Xu, J. Phys. Chem. B, 2006, 110, 4291–4300 CrossRef CAS PubMed.
- R. M. Yuan, G. Fu, X. Xu and H. L. Wan, Phys. Chem. Chem. Phys., 2011, 13, 453–460 RSC.
- H. Q. Wang, S. Gao, F. X. Yu, Y. Liu, X. L. Weng and Z. B. Wu, J. Phys. Chem. C, 2015, 119, 15077–15084 CAS.
- J. Valyon and W. K. Hall, J. Catal., 1993, 143, 520–532 CrossRef CAS.
- M. Iwamoto, H. Yahiro, N. Mizuno, W. X. Zhang, Y. Mine, H. Furukawa and S. Kagawa, J. Phys. Chem., 1992, 96, 9360–9366 CrossRef CAS.
- Z. Schay, H. Knozinger, L. Guczi and G. Pál-Borbély, Appl. Catal., B, 1998, 18, 263–271 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2016 |