
Silver-catalyzed synthesis of 2-arylvinylphosphonates by cross-coupling of β-nitrostyrenes with H-phosphites†
Jin-Wei Yuan*,
Liang-Ru Yang,
Pu Mao and
Ling-Bo Qu*
School of Chemistry & Chemical Engineering, Henan University of Technology, Academician Workstation for Natural Medicinal Chemistry of Henan Province, Zhengzhou 450001, P. R. China. E-mail: yuanjinweigs@126.com; Fax: +86-371-67756718; Tel: +86-371-67756718
First published on 8th September 2016
Abstract
An efficient protocol for stereoselective synthesis of 2-arylvinylphosphonates has been developed via AgNO3-catalyzed cross-coupling of β-nitrostyrenes with dialkyl H-phosphites under mild conditions. By losing the nitro group of β-nitrostyrenes, the reaction proceeds smoothly and could provide the desired products with moderate to good yield.
Introduction
Vinylphosphonates are important molecules known for their interesting properties spanning across chemistry as well as biology.1 They are suitable substrates for various named reactions such as Michael additions, cycloaddition, and Horner–Wadsworth–Emmons reactions.2 They have also been extensively used in polymer science as additives or flame-retardants.3 In medical chemistry, vinylphosphonates often exhibit interesting biological properties such as antiviral, antibacterial, and anticancer effects.4 For these reasons, the development of simple and stereoselective methods to prepare vinylphosphonates has become important in organic synthesis.In recent years, a wide variety of transition metal catalyzed cross-coupling reactions for the construction of C–P bond have been developed.5 As a significant motif in organic chemistry, the alkenyl Csp2–P bond formation has attracted much attention. There are two main strategies applicable in forming the alkenyl Csp2–P bond: the phosphorylation of alkynes or terminal alkynes, and functionalized alkenes. Dialkyl H-phosphites are coupled with functionalized alkenes including styrene, 2,2-dibromostyrenes, alkenyl acids, vinyliodonium tetrafluoroborates to form 2-arylvinylphosphonates using Cu as a catalyst.6 Transition-metals such as Ni, and Ni/Zn-catalyzed reactions of alkenyl acids, 2,2-dibromostyrenes with dialkyl phosphites have been reported.7 In 2015, Zou has described that Mn(III)-mediated alkenyl C(sp2)–P bond formation from the reaction of 2-nitrostyrenes with dialkyl phosphites, and Tan found an efficient Ag-catalyzed reaction of styrenes with dialkyl phosphites using K2S2O8 as the oxidant, and TEMPO as the additive8 (Scheme 1a). Transition metals such as Zr/Cu, Pd and Rh catalysts were employed for the reaction of terminal alkynes with Dialkyl H-phosphites and dialkyl chlorophosphates (Scheme 1b).9 It has also been reported that trialkyl phosphates react with 1-bromoalkenes and vinylboronate esters catalyzed by transition metal Pd, and Cu (Scheme 1c).10 Moreover, 2-arylvinylphosphonates are formed by the Mizoroki–Heck reaction of arylboronic acids with dialkyl vinylphosphonates catalyzed by Pd(OAc)2, and via nucleophilic substitution of benzyl bromides with Bestmann–Ohira reagents.11 Although these approaches are available for the synthesis of 2-arylvinylphosphonates, most of them suffer from several drawbacks such as lack of stereoselectivity (a mixture of E/Z products), the need to use a rather expensive catalyst, drastic conditions being not compatible with molecules containing sensitive functional groups. As a result, there is still a strong need for alternative methods that would allow the stereoselective synthesis of 2-arylvinylphosphonates from readily available starting materials and catalysts under mild reaction conditions.
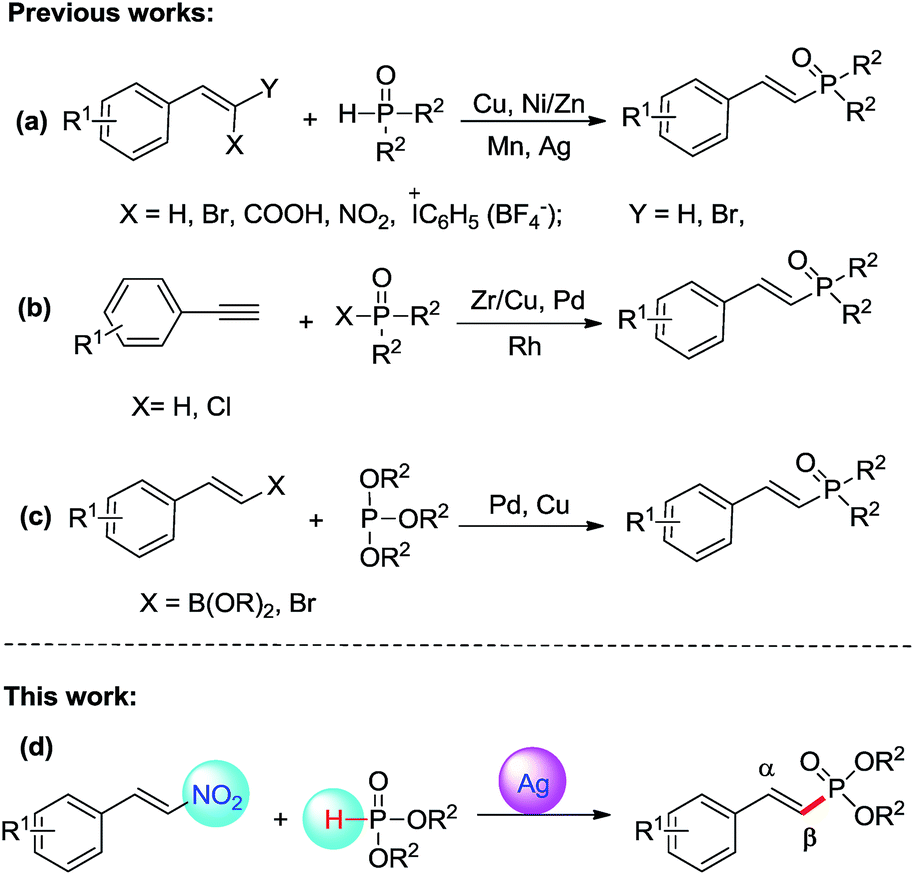 | ||
| Scheme 1 Strategies for synthesis of 2-arylvinylphosphonates. | ||
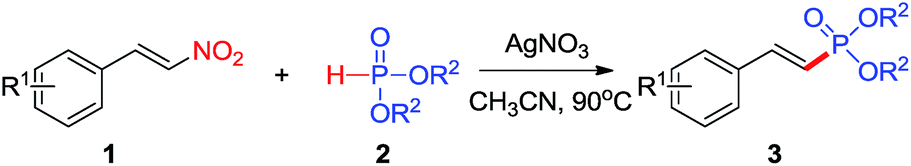
(E)-β-Nitrostyrenes are useful intermediates in organic synthesis and are important structural units that can be used as starting material for many classes of compounds.12 In general, the nitro group activates α- and/or β-position of a substrate, and in the reaction the nitro group is either remaining or leaving.13 (E)-β-Nitrostyrenes are good radical acceptors and react with alkyl radicals from different sources to generate (E)-alkenes under a variety of conditions and the reaction mechanism appears to involve a free-radical addition–elimination reaction.14 Although Zou has described that 2-arylvinylphosphonates could be synthsized by Mn(III)-catalyzed reaction of 2-nitrostyrenes with dialkyl phosphites,8a and Tan has found that Ag-catalyzed reaction of styrenes with dialkyl phosphites,8b these methodologies need use expensive and unstable catalysts, acid circumstance, strong oxidants, or with low yield. Drawing inspiration from recent studies15 that dialkyl phosphites could be utilized as a phosphoryl radical precursor,16 we wonder that 2-arylvinylphosphonates can be synthesized by cross-coupling of β-nitrostyrenes with dialkyl H-phosphites. Herein, we describe a new silver-catalyzed cross-coupling reaction between readily available β-nitrostyrenes with dialkyl H-phosphites, leading to 2-arylvinylphosphonates in moderate to excellent yields. Some notable features of this protocol are high efficiency, wide functional group tolerance, readily available and stable β-nitrostyrenes with dialkyl H-phosphites as starting materials, no oxidants and high stereoselectivity (Scheme 1d).
Results and discussion
Our previous studies showed that the phosphoryl radicals could be easily generated from H-phosphites with a catalytic amount of silver salts.16 Based on this achievement, the model reaction of (E)-β-nitrostyrenes (1a) and diisopropyl H-phosphite (2a) was carried out in the presence of AgNO3 (0.1 eq.) in CH3CN at 90 °C for 20 min. Much to our delight, the desired product (3a) was indeed obtained, and the isolated yield was 56% (Table 1, entry 1). The E or Z stereochemistry of 3a is easily established by the 1H NMR analysis. Indeed, the report in the literature has shown that the chemistry shift of β–H proton is 6.10–6.30 ppm with a coupling constant 17.0 Hz typical of trans positioned protons, while that of cis isomer gives 5.60–5.80 ppm with a coupling constant 15.0 Hz.9a The β-proton of 3a at δH = 6.27 ppm appears as a triplet with a coupling constant, J = 17.6 Hz, which proves that configuration of 3a is an E stereoisomer.
|
|||||
|---|---|---|---|---|---|
| Entry | Oxidant (eq.) | Solvent | Temp (°C) | Time (min) | Yieldb (%) |
| a Reaction conditions: (E)-β-nitrostyrene 1a (0.3 mmol, 44.7 mg), diisopropyl H-phosphite 2a (0.45 mmol, 74.7 mg), AgNO3 as the catalyst in solvent (3.0 mL).b Isolated yield.c nr = no reaction. | |||||
| 1 | AgNO3 (0.1) | CH3CN | 90 | 20 | 56 |
| 2 | AgNO3 (0.1) | H2O | 90 | 20 | <5 |
| 3 | AgNO3 (0.1) | Dioxane | 90 | 20 | 0 |
| 4 | AgNO3 (0.1) | CH3OH | 90 | 20 | 40 |
| 5 | AgNO3 (0.1) | DMSO | 90 | 20 | <5 |
| 6 | AgNO3 (0.1) | DCE | 90 | 20 | 54 |
| 7 | AgNO3 (0.1) | THF | 90 | 20 | 30 |
| 8 | AgNO3 (0.05) | CH3CN | 90 | 20 | 45 |
| 9 | AgNO3 (0.15) | CH3CN | 90 | 20 | 60 |
| 10 | AgNO3 (0.2) | CH3CN | 90 | 20 | 55 |
| 11 | AgNO3 (0.15) | CH3CN | 40 | 20 | Trace |
| 12 | AgNO3 (0.15) | CH3CN | 60 | 20 | 44 |
| 13 | AgNO3 (0.15) | CH3CN | 80 | 20 | 49 |
| 14 | AgNO3 (0.15) | CH3CN | 100 | 20 | 55 |
| 15 | AgOTf (0.15) | CH3CN | 90 | 20 | <5 |
| 16 | Ag2CO3 (0.15) | CH3CN | 90 | 20 | <5 |
| 17 | AgNO3 (0.15) | CH3CN | 90 | 40 | 65 |
| 18 | AgNO3 (0.15) | CH3CN | 90 | 1.0 h | 70 |
| 19 | AgNO3 (0.15) | CH3CN | 90 | 2.0 h | 78 |
| 20 | AgNO3 (0.15) | CH3CN | 90 | 3.0 h | 76 |
| 21 | — | CH3CN | 90 | 1.0 h | nrc |
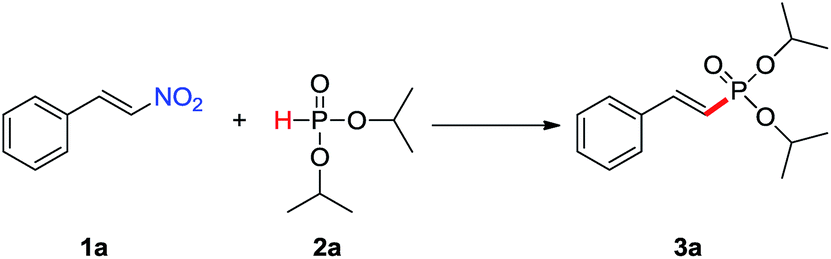
To achieve the optimal conditions, a variety of reaction conditions were employed. Initial screening of different solvents including H2O, dioxane, CH3OH, DMSO, DCE and THF were applied instead of CH3CN. The result revealed that solvents such as H2O and CH3OH are not suitable for this reaction, and CH3CN was clearly the best choice (Table 1, entries 1–7). The ratio of substrates (E)-β-nitrostyrenes and diisopropyl H-phosphite was investigated, and the ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.5 could provide the best result (Table S1, ESI†). The screening of the amount of the catalyst AgNO3 showed that a good yield (60%) of the product 3a was obtained when 0.15 equiv. of AgNO3 was employed, and excessive or less amount of the catalyst caused decreased yield (Table 1, entries 1, 8–10). Furthermore, various reaction temperatures were investigated, and increasing the temperature from 40 to 100 °C could enhance the reaction efficacy, and a good yield of 56% could be obtained at 90 °C (Table 1, entries 1, 11–14). Based on these joyful results, various catalysts of silver salts such as AgOTf and Ag2CO3 were further investigated. It was found that they proved to be less effective compared with AgNO3 (Table 1, entries 1, 15 and 16). In addition, various reaction times were also examined. The yield of 3a dramatically increased if the reaction time was increased, 2.0 h was found to be appropriate choice and the yield was 78% (Table 1, entries 1, 17–20). In the absence of AgNO3, the desired product 3a was not produced, which indicated that AgNO3 as a catalyst played an important role in this transformation (Table 1, entry 21).
:1.5 could provide the best result (Table S1, ESI†). The screening of the amount of the catalyst AgNO3 showed that a good yield (60%) of the product 3a was obtained when 0.15 equiv. of AgNO3 was employed, and excessive or less amount of the catalyst caused decreased yield (Table 1, entries 1, 8–10). Furthermore, various reaction temperatures were investigated, and increasing the temperature from 40 to 100 °C could enhance the reaction efficacy, and a good yield of 56% could be obtained at 90 °C (Table 1, entries 1, 11–14). Based on these joyful results, various catalysts of silver salts such as AgOTf and Ag2CO3 were further investigated. It was found that they proved to be less effective compared with AgNO3 (Table 1, entries 1, 15 and 16). In addition, various reaction times were also examined. The yield of 3a dramatically increased if the reaction time was increased, 2.0 h was found to be appropriate choice and the yield was 78% (Table 1, entries 1, 17–20). In the absence of AgNO3, the desired product 3a was not produced, which indicated that AgNO3 as a catalyst played an important role in this transformation (Table 1, entry 21).
With the optimized conditions in hand, we next set out to examine the scope of β-nitrostyrenes and H-phosphites, and the results are summarized in Table 2. A range of β-nitrostyrene derivatives were found to undergo denitration phosphonation in good to excellent yields ranging from 48–91% with high stereoselectivity (Table 2, entries 3a–3l, 3m–3zz). Substituents such as methyl, methoxy, methylenedioxy, fluoro, chloro, bromo, and iodo groups are well tolerated on the aromatic ring and their reactions afforded the target products in good to excellent yields, showing the broad scope of this reaction. β-Nitrostyrenes with electron-donating groups (Table 2, entries 3b–3e) could give slightly better yields than analogues with electron-withdrawing groups (Table 2, entries 3f–3j). The results indicated that electron-donating groups on the phenyl ring contributed to β-position carbon electron density, which made it more susceptible to an electrophilic attack by the phosphonyl radical. Notably, sterically demanding substrates like (E)-3,4-dimethoxyl-β-nitrostyrenes and (E)-2,6-dichloro-β-nitrostyrenes could be phosphorylated in 91% and 70% yields (Table 2, entries 3d and 3g). However, steric hindrance will lead to lower yield when β-methyl-β-nitrostyrene is employed (Table 2, entry 3l). It was gratifying to find that heterocyclic β-nitroalkene ((E)-2-(2-nitrovinyl)thiophene) also reacted smoothly with diisopropyl H-phosphite, leading to the corresponding product 3k in moderated yield (65%). Unfortunately, aliphatic nitroalkenes failed to deliver the desired products with the current catalytic system.
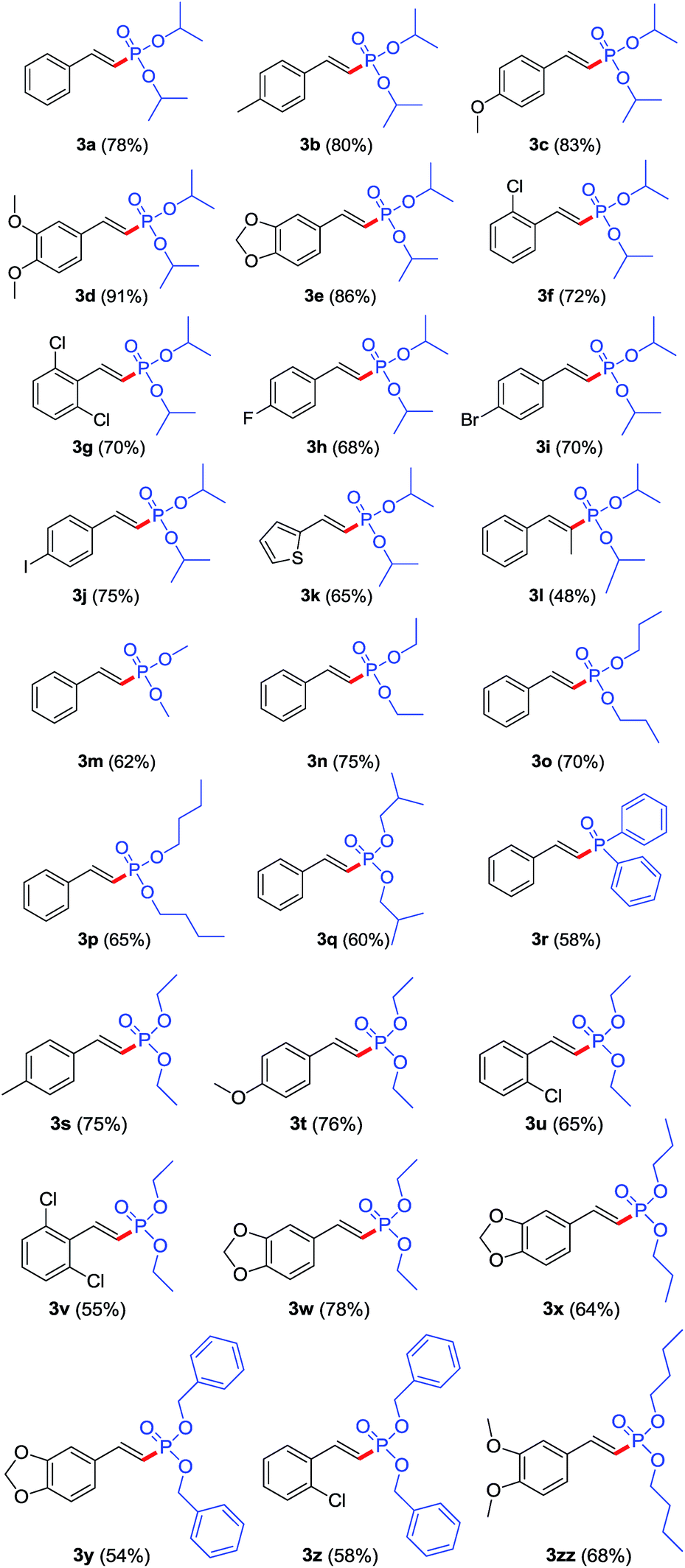
Subsequently, the scope for various dialkyl H-phosphites was also investigated. The reaction could proceed smoothly using different dialkyl H-phosphites to form the desired products in moderate to excellent yields in 60–78% with high stereoselectivity (Table 2, entries 3a, 3m–3q). It is gratifying to see that the reaction not only worked with dialkyl H-phosphites but also with diphenylphosphine oxide as well (Table 2, entry 3r). Using the standard reaction condition, the corresponding products were produced in moderate to good yields (Table 2, entries 3s–3zz). However, the yields of products 3y and 3z were lower when dibenzyl H-phosphite was employed, which was possible that partial product was hydrolyzed. Bisphosphonate drugs are used to treat a variety of bone resorption diseases, such as osteoporosis, Paget's disease, and hypercalcemia due to malignancy.17 In the study of using (E)-5-(2-nitrovinyl)benzo[d][1,3]dioxole and diethyl H-phosphite as the coupling partners, we found that this resulted in a mixture of mono-3w (yield: 50%) and bis-phosphorylated products 3w′ (yield: 24%) when 3.0 eq. diethyl H-phosphite was employed at 90 °C for 10 h (Scheme 2).
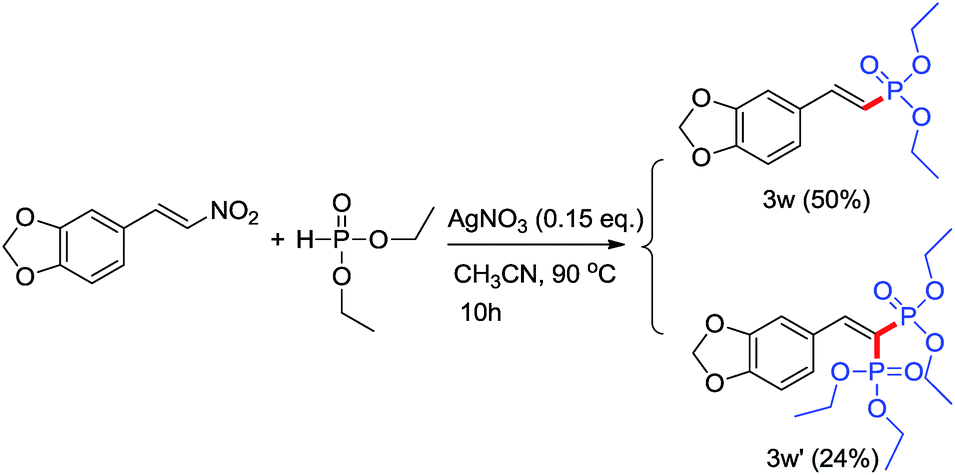 | ||
| Scheme 2 Synthesis of mono- and bis-phosphorylated products. | ||
To clarify the reaction mechanism, some controlled experiments were designed to investigate this transformation (Scheme 3). When the reaction of (E)-β-nitrostyrene 1a with diisopropyl H-phosphite 2a was performed under the standard conditions by the addition of 3.0 eqivalents radical scavengers, such as TEMPO and BHT, and the target product 3a decreased dramatically. These results indicated that the reaction might proceed via a radical pathway.
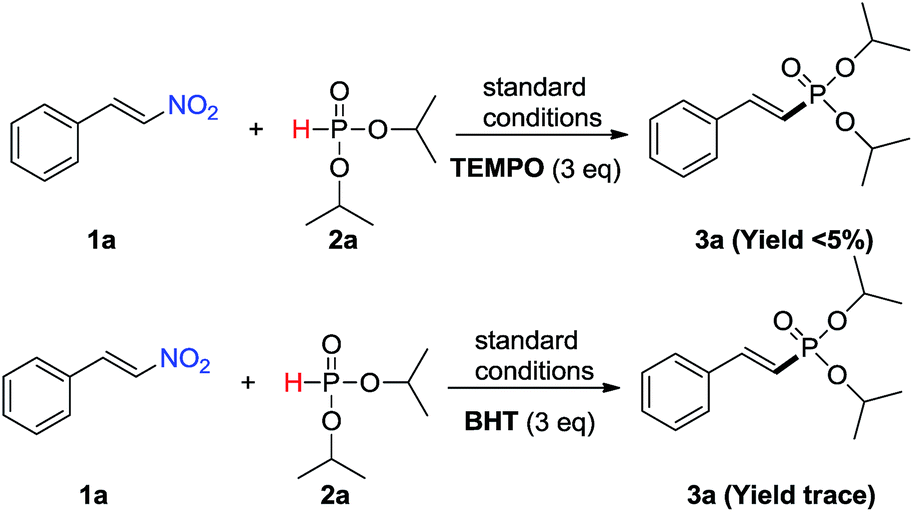 | ||
| Scheme 3 Controlled experiments. | ||
Previous works have shown that silver salts can react with H-phosphites to form the active (RO)2P(O)Ag complexes which subsequently generate the phosphoryl radical.16 Based on our controlled experimental results and literature precedents,8b,18 the following plausible mechanism can be proposed for the transformation (Scheme 4). The phosphoryl radical C may be generated from the complex B which itself is formed by the reaction of AgNO3 catalyst with H-phosphites A. Subsequently, the radical C selectively adds to the β-position of (E)-β-nitrostyrene to form a carbon-centered radical D, which undergoes an elimination reaction by the leaving NO2 radical to form the desired product E. Ag(0) could be oxidized back to Ag(I) by HNO3,16a,18a thus closing the catalytic cycle.
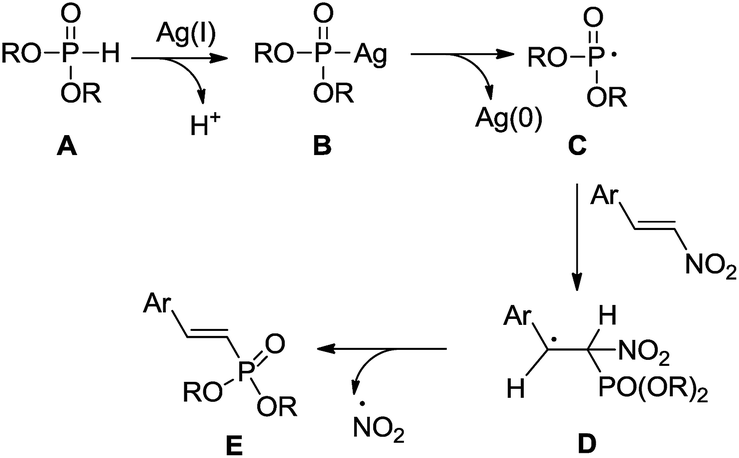 | ||
| Scheme 4 Proposed reaction mechanism for the formation of 2-arylvinylphosphonates. | ||
Conclusions
In conclusion, we have developed a novel and highly stereoselectivity protocol for the synthesis of 2-arylvinylphosphonates starting from (E)-β-nitrostyrene through a tandem radical addition-denitration process. This process features a broad substrate scope and functional group tolerance. The reaction proceeded under mild conditions to afford the selective (E)-2-arylvinylphosphonates in moderate to good yields.Experimental
General information
Anhydrous solvents were obtained by standard procedure. All substrates purchased from J & K Scientific Ltd. were used without further purification. Column chromatography was performed using 300–400 mesh silica with the indicated solvent system according to standard techniques. Nuclear magnetic resonance spectra were recorded on Bruker Avance 400 MHz spectrometer. Chemical shifts for 1H NMR spectra are recorded in parts per million from tetramethylsilane. Data were reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, m = multiplet and br = broad), coupling constant in Hz and integration. Chemical shifts for 13C NMR spectra were recorded in parts per million from tetramethylsilane. Chemical shift for 31P NMR spectra are recorded relative to 85% H3PO4 (δ = 0 ppm) as external standard. High resolution mass spectra (HR MS) were obtained on Q-TOF instrument using the ESI technique. IR spectra were recorded on Shimadazu IR-408 Fourier transform infrared spectrophotometer using a thin film supported on KBr pellets. Melting points were measured on an XT4A microscopic apparatus uncorrected.General procedure for synthesis of 2-arylvinylphosphonates (3)
(E)-β-Nitrostyrenes 1 (0.3 mmol), dialkyl H-phosphites 2 (0.45 mmol), and AgNO3 (0.045 mmol, 7.6 mg) in CH3CN (3.0 mL) were added to a 25 mL Schlenk tube. The mixture was heated at 90 °C for 2.0 h (monitored by TLC). After completion of the reaction, the solvent was distilled under vacuum. 10 mL ethylacetate was added to the residuum, 15 mL 5% NaHCO3 was added to wash two times, and 10 mL saturated NaCl solution washed one time. The organic phase was dried over anhydrous NaSO4 and concentrated under vacuum. The crude product was purified by silica gel column chromatography to give the desired products 3 using ethyl acetate/petroleum ether (1:3 to 2:1) as eluant.
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1105 (P–O). 1H NMR (CDCl3) δ: 7.53–7.43 (m, 3H), 7.39–7.31 (m, 3H), 6.27 (t, J = 17.6 Hz, 1H), 4.75–4.67 (m, 2H), 1.36 (d, J = 6.2 Hz, 6H), 1.31 (d, J = 6.2 Hz, 6H). 13C NMR (CDCl3) δ: 147.8 (d, JP–C = 6.6 Hz), 134.9 (d, JP–C = 22.9 Hz), 130.0, 128.7, 127.6, 115.5 (d, JP–C = 190.9 Hz), 70.4 (d, JP–C = 5.5 Hz), 24.0 (d, JP–C = 4.1 Hz), 23.9 (d, JP–C = 4.8 Hz). 31P NMR (CDCl3) δ: 17.4. MS (ESI) m/z: 269.3 [M + H]+ (calcd for C14H22O3P+ 269.1).O), 1107 (P–O). 1H NMR (CDCl3) δ: 7.46 (dd, J = 22.6 Hz, J = 17.4 Hz, 1H), 7.39 (d, J = 8.0 Hz, 2H), 7.18 (d, J = 8.0 Hz, 2H), 6.20 (t, J = 17.5 Hz, 1H), 4.75–4.66 (m, 2H), 2.36 (s, 3H), 1.36 (d, J = 6.2 Hz, 6H), 1.31 (d, J = 6.2 Hz, 6H). 13C NMR (CDCl3) δ: 147.8 (d, JP–C = 6.7 Hz), 140.4, 132.3 (d, JP–C = 23.3 Hz), 129.5, 127.6, 114.2 (d, JP–C = 191.5 Hz), 70.4 (d, JP–C = 5.6 Hz), 24.1 (d, JP–C = 4.1 Hz), 24.0 (d, JP–C = 4.7 Hz), 21.4. 31P NMR (CDCl3) δ: 17.8. MS (ESI) m/z: 283.2 [M + H]+ (calcd for C15H24O3P+ 283.1).O), 1103 (P–O). 1H NMR (CDCl3) δ: 7.49–7.32 (m, 3H), 6.89 (d, J = 8.7 Hz, 2H), 6.10 (t, J = 17.4 Hz, 1H), 4.74–4.66 (m, 2H), 3.82 (s, 3H), 1.36 (d, J = 6.2 Hz, 6H), 1.31 (d, J = 6.2 Hz, 6H). 13C NMR (CDCl3) δ: 161.1, 147.5 (d, JP–C = 6.8 Hz), 129.2, 127.7 (d, JP–C = 23.5 Hz), 114.1, 112.2 (d, JP–C = 192.4 Hz), 70.3 (d, JP–C = 5.4 Hz), 55.3, 24.0 (d, JP–C = 4.0 Hz), 23.9 (d, JP–C = 4.5 Hz). 31P NMR (CDCl3) δ: 18.3. MS (ESI) m/z: 299.2 [M + H]+ (calcd for C15H24O4P+ 299.1).O), 1074 (P–O). 1H NMR (CDCl3) δ: 7.36 (dd, J = 22.5 Hz, J = 17.4 Hz, 1H), 7.03 (dd, J = 8.2 Hz, J = 1.8 Hz, 1H), 6.98 (d, J = 1.8 Hz, 1H), 6.81 (d, J = 8.3 Hz, 1H), 6.06 (t, J = 17.4 Hz, 1H), 4.70–4.62 (m, 2H), 3.86 (s, 6H), 1.32 (d, J = 6.2 Hz, 6H), 1.28 (d, J = 6.2 Hz, 6H). 13C NMR (CDCl3) δ: 150.8, 149.1, 147.6 (d, JP–C = 6.9 Hz, CH), 128.0 (d, JP–C = 23.5 Hz), 121.9 (CH), 112.8 (d, JP–C = 192.5 Hz, CH), 110.9 (CH), 109.3 (CH), 70.3 (d, JP–C = 5.4 Hz), 55.9 (CH3), 55.8 (CH3), 24.1 (d, JP–C = 4.2 Hz, CH3), 24.0 (d, JP–C = 4.2 Hz, CH3). 31P NMR (CDCl3) δ: 18.0. MS (ESI) m/z: 329.2 [M + H]+ (calcd for C16H26O5P+ 329.1).O), 1105 (P–O). 1H NMR (CDCl3) δ: 7.37 (dd, J = 22.4 Hz, J = 17.4 Hz, 1H), 7.00 (d, J = 1.1 Hz, 1H), 6.96 (d, J = 9.3 Hz, 1H), 6.79 (d, J = 8.0 Hz, 1H), 6.07 (t, J = 17.2 Hz, 1H), 5.99 (s, 2H), 4.74–4.65 (m, 2H), 1.36 (d, J = 6.2 Hz, 6H), 1.31 (d, J = 6.2 Hz, 6H). 13C NMR (CDCl3) δ: 149.3, 148.2, 147.3 (d, JP–C = 7.2 Hz, CH), 129.4 (d, JP–C = 23.8 Hz), 123.7 (CH), 113.0 (d, JP–C = 192.1 Hz, CH), 107.2 (d, JP–C = 225.5 Hz, CH), 101.4 (CH), 70.3 (d, JP–C = 5.5 Hz), 24.0 (d, JP–C = 4.0 Hz, CH3), 23.9 (d, JP–C = 4.2 Hz, CH3). 31P NMR (CDCl3) δ: 17.8. MS (ESI) m/z: 313.3 [M + H]+ (calcd for C15H22O5P+ 313.1).O), 1105 (P–O), 754 (C–Cl). 1H NMR (CDCl3) δ: 7.83 (dd, J = 22.6 Hz, J = 17.5 Hz, 1H), 7.60–7.57 (m, 1H), 7.40–7.38 (m, 1H), 7.31–7.27 (m, 2H), 6.31 (t, J = 17.6 Hz, 1H), 4.79–4.70 (m, 2H), 1.36 (dd, J = 12.7 Hz, J = 6.2 Hz, 12H). 13C NMR (CDCl3) δ: 143.0 (d, JP–C = 7.8 Hz), 134.4, 133.2 (d, JP–C = 23.7 Hz), 130.8, 130.0 (d, JP–C = 1.0 Hz), 127.3, 127.0, 119.8, 117.9, 70.7 (d, JP–C = 5.7 Hz), 24.1 (d, JP–C = 4.0 Hz), 24.0 (d, JP–C = 4.8 Hz). 31P NMR (CDCl3) δ: 16.0. HR MS (ESI) m/z: 303.0914 [M + H]+ (calcd for C14H21ClO3P+ 303.0911).O), 1153 (P–O), 756 (C–Cl). 1H NMR (CDCl3) δ: 7.51 (dd, J = 23.6 Hz, J = 17.8 Hz, 1H), 7.34 (d, J = 8.0 Hz, 2H), 7.18 (t, J = 8.0 Hz, 1H), 6.43 (t, J = 18.0 Hz, 1H), 4.80–4.72 (m, 2H), 1.38 (d, J = 6.2 Hz, 6H), 1.31 (d, J = 6.2 Hz, 6H). 13C NMR (CDCl3) δ: 141.3 (d, JP–C = 7.8 Hz), 134.5 (d, JP–C = 1.5 Hz), 132.8 (d, JP–C = 23.4 Hz), 129.6, 128.7, 125.8 (d, JP–C = 183.2 Hz), 70.5 (d, JP–C = 5.4 Hz), 24.1 (d, JP–C = 4.0 Hz), 23.9 (d, JP–C = 4.5 Hz). 31P NMR (CDCl3) δ: 14.6. HR MS (ESI) m/z: 337.0518 [M + H]+ (calcd for C14H20Cl2O3P+ 337.0522).O), 1108 (P–O). 1H NMR (CDCl3) δ: 7.45–7.34 (m, 3H), 7.02 (d, J = 8.6 Hz, 2H), 6.14 (t, J = 17.2 Hz, 1H), 4.71–4.63 (m, 2H), 1.32 (d, J = 6.2 Hz, 6H), 1.28 (d, J = 6.2 Hz, 6H). 13C NMR (CDCl3) δ: 163.7 (d, JF–C = 249.1 Hz), 146.4 (d, JP–C = 6.9 Hz, CH), 131.2 (dd, JF–C = 3.4 Hz, JP–C = 23.5 Hz), 129.4 (d, JP–C = 8.4 Hz, CH), 115.8 (d, JP–C = 21.8 Hz, CH), 115.3 (dd, JF–C = 2.0 Hz, JP–C = 191.7 Hz, CH), 70.5 (d, JP–C = 5.5 Hz, CH), 24.0 (d, JP–C = 4.0 Hz, CH3), 23.9 (d, JP–C = 4.0 Hz, CH3). 31P NMR (CDCl3) δ: 17.0. 19F NMR (CDCl3) δ: −110.2. MS (ESI) m/z: 287.3 [M + H]+ (calcd for C14H21FO3P+ 287.1).O), 1116 (P–O). 1H NMR (CDCl3) δ: 7.51–7.39 (m, 3H), 7.34 (d, J = 8.4 Hz, 2H), 6.24 (t, J = 17.2 Hz, 1H), 4.76–4.66 (m, 2H), 1.35 (d, J = 6.2 Hz, 6H), 1.30 (d, J = 6.2 Hz, 6H). 13C NMR (CDCl3) δ: 146.4 (d, JP–C = 7.0 Hz), 133.9 (d, JP–C = 23.5 Hz), 132.0, 129.0, 124.2, 116.2 (d, JP–C = 191.5 Hz), 70.5 (d, JP–C = 5.8 Hz), 24.0 (d, JP–C = 4.0 Hz), 23.9 (d, JP–C = 4.5 Hz). 31P NMR (CDCl3) δ: 16.6. MS (ESI) m/z: 347.2 [M + H]+ (calcd for C14H21BrO3P+ 347.0).O), 1109 (P–O). 1H NMR (CDCl3) δ: 7.70 (d, J = 8.4 Hz, 2H), 7.37 (dd, J = 17.6 Hz, J = 22.6 Hz, 1H), 7.20 (d, J = 8.4 Hz, 2H), 6.25 (t, J = 17.2 Hz, 1H), 4.73–4.65 (m, 2H), 1.35 (d, J = 6.2 Hz, 6H), 1.30 (d, J = 6.2 Hz, 6H). 13C NMR (CDCl3) δ: 146.4 (d, JP–C = 6.7 Hz), 138.0, 134.5 (d, JP–C = 23.3 Hz), 129.1, 127.6, 116.6 (d, JP–C = 191.0 Hz), 70.6 (d, JP–C = 5.7 Hz), 24.1 (d, JP–C = 4.0 Hz), 24.0 (d, JP–C = 4.5 Hz). 31P NMR (CDCl3) δ: 16.6. HR MS (ESI) m/z: 395.0270 [M + H]+ (calcd for C14H21IO3P+ 395.0268).O), 1103 (P–O). 1H NMR (CDCl3) δ: 7.49 (dd, J = 22.6 Hz, J = 17.4 Hz, 1H), 7.28 (d, J = 5.0 Hz, 1H), 7.13 (d, J = 3.3 Hz, 1H), 6.97 (t, J = 4.6 Hz, 1H), 5.95 (t, J = 16.8 Hz, 1H), 4.69–4.59 (m, 2H), 1.30 (d, J = 6.2 Hz, 6H), 1.26 (d, J = 6.2 Hz, 6H). 13C NMR (CDCl3) δ: 140.5 (d, JP–C = 26.7 Hz), 140.2 (d, JP–C = 7.6 Hz, CH), 129.9 (CH), 127.9 (d, JP–C = 1.1 Hz, CH), 127.8, 114.1 (d, JP–C = 193.1 Hz, CH), 70.5 (d, JP–C = 5.5 Hz, CH), 24.0 (d, JP–C = 4.0 Hz, CH3), 23.9 (d, JP–C = 4.0 Hz, CH3). 31P NMR (CDCl3) δ: 16.7. MS (ESI) m/z: 275.2 [M + H]+ (calcd for C12H20O3PS+ 275.0).O), 1132 (P–O). 1H NMR (CDCl3) δ: 7.47 (dd, J = 24.8 Hz, J = 1.4 Hz, 1H), 7.38 (d, J = 4.4 Hz, 4H), 7.34–7.29 (m, 1H), 4.71–4.68 (m, 2H), 2.06 (dd, J = 15.2 Hz, J = 1.4 Hz, 1H), 1.38 (d, J = 6.2 Hz, 6H), 1.31 (d, J = 6.2 Hz, 6H). 13C NMR (CDCl3) δ: 141.9 (d, JP–C = 12.5 Hz, CH), 135.9 (d, JP–C = 23.4 Hz), 129.4 (CH), 128.4 (CH), 128.2 (CH), 126.4, 70.3 (d, JP–C = 6.4 Hz, CH), 24.1 (d, JP–C = 4.2 Hz, CH3), 23.8 (d, JP–C = 4.5 Hz, CH3), 14.4 (d, JP–C = 9.7 Hz, CH3). 31P NMR (CDCl3) δ: 19.7. HR MS (ESI) m/z: 283.1456 [M + H]+ (calcd for C15H24O3P+ 283.1458).O), 1053 (P–O). 1H NMR (CDCl3) δ: 7.58–7.48 (m, 3H), 7.40–7.37 (m, 3H), 6.23 (t, J = 17.7 Hz, 1H), 3.78 (d, J = 11.1 Hz, 6H). 13C NMR (CDCl3) δ: 149.6 (d, JP–C = 6.6 Hz), 134.6 (d, JP–C = 23.1 Hz), 130.4, 128.9, 127.7, 112.3 (d, JP–C = 191.2 Hz), 52.4 (d, JP–C = 5.6 Hz). 31P NMR (CDCl3) δ: 22.4. MS (ESI) m/z: 213.1 [M + H]+ (calcd for C10H14O3P+ 213.0).O), 1134 (P–O). 1H NMR (CDCl3) δ: 7.56–7.46 (m, 3H), 7.40–7.37 (m, 3H), 6.26 (t, J = 17.6 Hz, 1H), 4.17–4.09 (m, 4H), 1.36 (t, J = 7.0 Hz, 6H). 13C NMR (CDCl3) δ: 148.7 (d, JP–C = 6.7 Hz), 134.8 (d, JP–C = 23.0 Hz), 130.2, 128.8, 127.7, 113.9 (d, JP–C = 190.3 Hz), 61.8 (d, JP–C = 5.5 Hz), 16.4 (d, JP–C = 6.4 Hz). 31P NMR (CDCl3) δ: 19.5. MS (ESI) m/z: 241.3 [M + H]+ (calcd for C12H18O3P+ 241.1).O), 1065 (P–O). 1H NMR (CDCl3) δ: 7.56–7.46 (m, 3H), 7.39–7.37 (m, 3H), 6.26 (t, J = 17.6 Hz, 1H), 4.05–3.99 (m, 4H), 1.76–1.68 (m, 4H), 0.97 (d, J = 7.4 Hz, 6H). 13C NMR (CDCl3) δ: 148.8 (d, JP–C = 6.7 Hz), 134.8 (d, JP–C = 23.2 Hz), 130.3, 128.9, 127.7, 113.6 (d, JP–C = 191.0 Hz), 67.4 (d, JP–C = 5.8 Hz), 23.8 (d, JP–C = 6.5 Hz), 10.0. 31P NMR (CDCl3) δ: 19.6. MS (ESI) m/z: 269.2 [M + H]+ (calcd for C14H22O3P+ 269.1).O), 1068 (P–O). 1H NMR (CDCl3) δ: 7.52–7.42 (m, 3H), 7.36–7.33 (m, 3H), 6.24 (t, J = 17.6 Hz, 1H), 4.06–3.98 (m, 4H), 1.67–1.60 (m, 4H), 1.43–1.35 (m, 4H), 0.89 (t, J = 7.4 Hz, 6H). 13C NMR (CDCl3) δ: 148.7 (d, JP–C = 6.6 Hz, CH), 134.8 (d, JP–C = 23.1 Hz), 130.2 (CH), 128.8 (CH), 127.7 (CH), 113.8 (d, JP–C = 190.4 Hz, CH), 65.5 (d, JP–C = 5.7 Hz, CH2), 32.5 (d, JP–C = 6.3 Hz, CH2), 18.7 (CH2), 13.6 (CH3). 31P NMR (CDCl3) δ: 19.6. MS (ESI) m/z: 297.3 [M + H]+ (calcd for C16H26O3P+ 297.1).O), 1047 (P–O). 1H NMR (CDCl3) δ: 7.57–7.47 (m, 3H), 7.39–7.37 (m, 3H), 6.26 (t, J = 17.6 Hz, 1H), 3.85–3.80 (m, 4H), 2.00–1.93 (m, 2H), 0.95 (d, J = 6.8 Hz, 9H). 13C NMR (CDCl3) δ: 148.9 (d, JP–C = 6.5 Hz), 134.8 (d, JP–C = 23.3 Hz), 130.3, 128.9, 127.7, 113.4 (d, JP–C = 191.7 Hz), 71.9 (d, JP–C = 6.1 Hz), 29.1 (d, JP–C = 6.7 Hz), 18.7 (d, JP–C = 1.9 Hz). 31P NMR (CDCl3) δ: 19.4. HR MS (ESI) m/z: 297.1618 [M + H]+ (calcd for C16H26O3P+ 297.1614).O), 1120 (P–O). 1H NMR (CDCl3) δ: 7.77–7.72 (m, 4H), 7.55–7.44 (m, 9H), 7.36–7.35 (m, 3H), 6.83 (dd, J = 22.4 Hz, J = 17.4 Hz, 1H). 13C NMR (CDCl3) δ: 147.6 (d, JP–C = 3.5 Hz, CH), 135.1 (d, JP–C = 17.8 Hz), 132.8 (d, JP–C = 105.5 Hz), 131.9 (d, JP–C = 2.5 Hz, CH), 131.4 (d, JP–C = 10.0 Hz, CH), 130.1, 128.8 (d, JP–C = 27.8 Hz, CH), 128.7 (CH), 127.8 (CH), 119.0 (d, JP–C = 103.9 Hz, CH). 31P NMR (CDCl3) δ: 24.8. MS (ESI) m/z: 305.0 [M + H]+ (calcd for C20H18OP+ 305.1).O), 1026 (P–O). 1H NMR (CDCl3) δ: 7.47 (dd, J = 22.6 Hz, J = 17.5 Hz, 1H), 7.39 (d, J = 8.1 Hz, 2H), 7.18 (d, J = 8.1 Hz, 2H), 6.19 (t, J = 17.7 Hz, 1H), 4.16–4.08 (m, 4H), 2.37 (s, 3H), 1.35 (t, J = 7.0 Hz, 6H). 13C NMR (CDCl3) δ: 148.8 (d, JP–C = 6.6 Hz), 140.6, 132.1 (d, JP–C = 23.2 Hz), 129.5, 127.7, 112.4 (d, JP–C = 190.8 Hz), 61.8 (d, JP–C = 5.4 Hz), 21.4, 16.4 (d, JP–C = 6.5 Hz). 31P NMR (CDCl3) δ: 19.9. MS (ESI) m/z: 255.0 [M + H]+ (calcd for C13H20O3P+ 255.1).O), 1028 (P–O). 1H NMR (CDCl3) δ: 7.46–7.36 (m, 3H), 6.86 (d, J = 8.8 Hz, 2H), 6.05 (t, J = 17.6 Hz, 1H), 4.12–4.04 (m, 4H), 3.79 (s, 3H), 1.31 (t, J = 7.1 Hz, 6H). 13C NMR (CDCl3) δ: 161.2, 148.4 (d, JP–C = 6.9 Hz, CH), 129.3 (CH), 127.6 (d, JP–C = 23.7 Hz), 114.2 (CH), 110.6 (d, JP–C = 191.7 Hz, CH), 61.7 (d, JP–C = 5.3 Hz, CH2), 55.3 (CH3), 16.4 (d, JP–C = 6.5 Hz, CH3). 31P NMR (CDCl3) δ: 20.4. MS (ESI) m/z: 271.3 [M + H]+ (calcd for C13H20O4P+ 271.1).O), 1075 (P–O), 806 (C–Cl). 1H NMR (CDCl3) δ: 7.86 (dd, J = 22.6 Hz, J = 17.5 Hz, 1H), 7.61–7.58 (m, 1H), 7.41–7.39 (m, 1H), 7.31–7.28 (m, 2H), 6.30 (t, J = 17.7 Hz, 1H), 4.20–4.12 (m, 4H), 1.37 (t, J = 7.0 Hz, 6H). 13C NMR (CDCl3) δ: 144.0 (d, JP–C = 7.8 Hz), 134.5, 133.2 (d, JP–C = 23.5 Hz), 131.0, 130.0, 127.4, 127.0, 117.2 (d, JP–C = 33.0 Hz), 62.1 (d, JP–C = 5.6 Hz), 16.4 (d, JP–C = 6.4 Hz). 31P NMR (CDCl3) δ: 18.2. MS (ESI) m/z: 275.2 [M + H]+ (calcd for C12H17ClO3P+ 275.0).O), 1151 (P–O), 764 (C–Cl). 1H NMR (CDCl3) δ: 7.53 (dd, J = 23.7 Hz, J = 17.9 Hz, 1H), 7.36 (d, J = 7.7 Hz, 2H), 7.19 (t, J = 8.2 Hz, 1H), 6.42 (t, J = 18.2 Hz, 1H), 4.21–4.14 (m, 4H), 1.38 (d, J = 7.0 Hz, 6H). 13C NMR (CDCl3) δ: 142.1 (d, JP–C = 7.6 Hz), 134.5 (d, JP–C = 1.4 Hz), 129.8, 128.7, 127.9, 124.2 (d, JP–C = 182.5 Hz), 62.2 (d, JP–C = 5.4 Hz), 16.4 (d, JP–C = 6.4 Hz). 31P NMR (CDCl3) δ: 17.0. MS (ESI) m/z: 309.2 [M + H]+ (calcd for C12H16Cl2O3P+ 309.0).O), 1038 (P–O). 1H NMR (CDCl3) δ: 7.33 (dd, J = 22.4 Hz, J = 17.4 Hz, 1H), 6.94 (s, 1H), 6.90 (d, J = 8.0 Hz, 1H), 6.73 (d, J = 8.0 Hz, 1H), 5.98 (t, J = 17.4 Hz, 1H), 5.93 (s, 2H), 4.09–4.01 (m, 4H), 1.28 (t, J = 7.1 Hz, 6H). 13C NMR (CDCl3) δ: 149.4, 148.4, 148.3 (d, JP–C = 3.8 Hz, CH), 129.3 (d, JP–C = 23.8 Hz), 123.9 (CH), 112.3 (CH), 110.4 (CH), 108.4 (CH), 106.2 (CH), 101.5 (CH2), 61.7 (d, JP–C = 5.5 Hz, CH2), 16.3 (d, JP–C = 6.4 Hz, CH3). 31P NMR (CDCl3) δ: 19.6. MS (ESI) m/z: 285.2 [M + H]+ (calcd for C13H18O5P+ 285.0).O), 1106 (P–O). 1H NMR (CDCl3) δ: 8.18 (dd, J = 29.4 Hz, J = 47.7 Hz, 1H), 7.55 (d, J = 1.3 Hz, 1H), 7.30 (d, J = 8.2 Hz, 1H), 6.83 (d, J = 8.2 Hz, 1H), 6.83 (s, 2H), 4.22–4.14 (m, 4H), 4.13–4.05 (m, 4H), 1.37 (t, J = 7.0 Hz, 6H), 1.24 (t, J = 7.0 Hz, 6H). 13C NMR (CDCl3) δ: 161.1 (d, JP–C = 2.2 Hz), 150.1, 147.6, 128.3 (dd, JP–C = 22.6 Hz, JP–C = 8.8 Hz), 127.8, 110.6, 107.9, 101.6, 62.6 (d, JP–C = 5.2 Hz), 62.4 (d, JP–C = 5.8 Hz), 16.3 (d, JP–C = 6.6 Hz), 16.1 (d, JP–C = 6.6 Hz). 31P NMR (CDCl3) δ: 18.3 (d, JP–P = 49.5 Hz), 12.8 (d, JP–P = 49.5 Hz). HR MS (ESI) m/z: 421.1178 [M + H]+ (calcd for C17H27O8P2+ 421.1176).O), 1036 (P–O). 1H NMR (CDCl3) δ: 7.40 (dd, J = 22.4 Hz, J = 17.4 Hz, 1H), 7.01 (d, J = 1.3 Hz, 1H), 6.97 (d, J = 8.0 Hz, 1H), 6.80 (d, J = 8.0 Hz, 1H), 6.09–6.00 (m, 3H), 4.03–3.97 (m, 4H), 1.76–1.67 (m, 4H), 0.96 (t, J = 7.4 Hz, 6H). 13C NMR (CDCl3) δ: 149.4, 148.3 (d, JP–C = 5.0 Hz), 129.3 (d, JP–C = 23.7 Hz), 123.9, 112.3, 110.4, 108.4, 106.2, 101.5, 67.3 (d, JP–C = 5.5 Hz), 23.8 (d, JP–C = 6.5 Hz), 10.1. 31P NMR (CDCl3) δ: 20.1. HR MS (ESI) m/z: 313.1202 [M + H]+ (calcd for C15H22O5P+ 313.1199).O), 1036 (P–O). 1H NMR (CDCl3) δ: 7.38–7.29 (m, 11H), 6.90–6.87 (m, 2H), 6.76 (d, J = 7.9 Hz, 1H), 6.01 (t, J = 17.6 Hz, 1H), 5.96 (s, 2H), 5.07 (s, 2H), 5.05 (s, 2H). 13C NMR (CDCl3) δ: 149.6, 148.7 (d, JP–C = 7.2 Hz, CH), 136.3 (d, JP–C = 6.6 Hz), 129.2 (d, JP–C = 24.4 Hz), 128.6 (CH), 128.4 (CH), 127.9 (CH), 124.0 (CH), 111.0 (d, JP–C = 192.8 Hz, CH), 108.4 (CH), 106.2 (CH), 101.5 (CH2), 67.4 (d, JP–C = 5.2 Hz, CH2). 31P NMR (CDCl3) δ: 21.1. HR MS (ESI) m/z: 409.1196 [M + H]+ (calcd for C23H22O5P+ 409.1199).O), 1049 (P–O). 1H NMR (CDCl3) δ: 7.87 (dd, J = 23.0 Hz, J = 17.5 Hz, 1H), 7.47 (dd, J = 7.5 Hz, J = 1.6 Hz, 1H), 7.39–7.24 (m, 13H), 6.24 (t, J = 18.1 Hz, 1H), 5.11 (d, J = 2.8 Hz, 2H), 5.09 (d, J = 2.7 Hz, 2H). 13C NMR (CDCl3) δ: 144.4 (d, JP–C = 8.1 Hz, CH), 136.1 (d, JP–C = 6.5 Hz), 134.6, 133.0 (d, JP–C = 23.9 Hz), 131.0 (CH), 130.1 (CH), 128.6 (CH), 128.4 (CH), 128.0 (CH), 127.4 (d, JP–C = 1.0 Hz, CH), 127.0 (CH), 116.9 (d, JP–C = 190.9 Hz, CH), 67.5 (d, JP–C = 5.6 Hz, CH2). 31P NMR (CDCl3) δ: 19.2. HR MS (ESI) m/z: 399.0909 [M + H]+ (calcd for C22H21ClO3P+ 399.0911).O), 1159 (P–O). 1H NMR (CDCl3) δ: 7.37 (dd, J = 22.4 Hz, J = 17.4 Hz, 1H), 7.02 (dd, J = 8.2 Hz, J = 1.5 Hz, 1H), 6.98 (d, J = 1.5 Hz, 1H), 6.81 (d, J = 8.3 Hz, 1H), 6.04 (t, J = 17.4 Hz, 1H), 4.03–3.96 (m, 4H), 3.85 (s, 6H), 1.66–1.59 (m, 4H), 1.41–1.32 (m, 4H), 0.87 (t, J = 7.4 Hz, 6H). 13C NMR (CDCl3) δ: 150.9, 149.1, 148.5 (d, JP–C = 6.9 Hz, CH), 127.8 (d, JP–C = 23.5 Hz), 122.0 (CH), 111.0 (d, JP–C = 191.9 Hz, CH), 110.9 (CH), 109.3 (CH), 65.5 (CH3), 65.4 (CH3), 55.9 (d, JP–C = 8.9 Hz, CH2), 32.5 (d, JP–C = 6.5 Hz, CH2), 18.7 (CH2), 13.6 (CH3). 31P NMR (CDCl3) δ: 19.8. HR MS (ESI) m/z: 357.1823 [M + H]+ (calcd for C18H30O5P+ 357.1825).
O), 1105 (P–O). 1H NMR (CDCl3) δ: 7.53–7.43 (m, 3H), 7.39–7.31 (m, 3H), 6.27 (t, J = 17.6 Hz, 1H), 4.75–4.67 (m, 2H), 1.36 (d, J = 6.2 Hz, 6H), 1.31 (d, J = 6.2 Hz, 6H). 13C NMR (CDCl3) δ: 147.8 (d, JP–C = 6.6 Hz), 134.9 (d, JP–C = 22.9 Hz), 130.0, 128.7, 127.6, 115.5 (d, JP–C = 190.9 Hz), 70.4 (d, JP–C = 5.5 Hz), 24.0 (d, JP–C = 4.1 Hz), 23.9 (d, JP–C = 4.8 Hz). 31P NMR (CDCl3) δ: 17.4. MS (ESI) m/z: 269.3 [M + H]+ (calcd for C14H22O3P+ 269.1).O), 1107 (P–O). 1H NMR (CDCl3) δ: 7.46 (dd, J = 22.6 Hz, J = 17.4 Hz, 1H), 7.39 (d, J = 8.0 Hz, 2H), 7.18 (d, J = 8.0 Hz, 2H), 6.20 (t, J = 17.5 Hz, 1H), 4.75–4.66 (m, 2H), 2.36 (s, 3H), 1.36 (d, J = 6.2 Hz, 6H), 1.31 (d, J = 6.2 Hz, 6H). 13C NMR (CDCl3) δ: 147.8 (d, JP–C = 6.7 Hz), 140.4, 132.3 (d, JP–C = 23.3 Hz), 129.5, 127.6, 114.2 (d, JP–C = 191.5 Hz), 70.4 (d, JP–C = 5.6 Hz), 24.1 (d, JP–C = 4.1 Hz), 24.0 (d, JP–C = 4.7 Hz), 21.4. 31P NMR (CDCl3) δ: 17.8. MS (ESI) m/z: 283.2 [M + H]+ (calcd for C15H24O3P+ 283.1).O), 1103 (P–O). 1H NMR (CDCl3) δ: 7.49–7.32 (m, 3H), 6.89 (d, J = 8.7 Hz, 2H), 6.10 (t, J = 17.4 Hz, 1H), 4.74–4.66 (m, 2H), 3.82 (s, 3H), 1.36 (d, J = 6.2 Hz, 6H), 1.31 (d, J = 6.2 Hz, 6H). 13C NMR (CDCl3) δ: 161.1, 147.5 (d, JP–C = 6.8 Hz), 129.2, 127.7 (d, JP–C = 23.5 Hz), 114.1, 112.2 (d, JP–C = 192.4 Hz), 70.3 (d, JP–C = 5.4 Hz), 55.3, 24.0 (d, JP–C = 4.0 Hz), 23.9 (d, JP–C = 4.5 Hz). 31P NMR (CDCl3) δ: 18.3. MS (ESI) m/z: 299.2 [M + H]+ (calcd for C15H24O4P+ 299.1).O), 1074 (P–O). 1H NMR (CDCl3) δ: 7.36 (dd, J = 22.5 Hz, J = 17.4 Hz, 1H), 7.03 (dd, J = 8.2 Hz, J = 1.8 Hz, 1H), 6.98 (d, J = 1.8 Hz, 1H), 6.81 (d, J = 8.3 Hz, 1H), 6.06 (t, J = 17.4 Hz, 1H), 4.70–4.62 (m, 2H), 3.86 (s, 6H), 1.32 (d, J = 6.2 Hz, 6H), 1.28 (d, J = 6.2 Hz, 6H). 13C NMR (CDCl3) δ: 150.8, 149.1, 147.6 (d, JP–C = 6.9 Hz, CH), 128.0 (d, JP–C = 23.5 Hz), 121.9 (CH), 112.8 (d, JP–C = 192.5 Hz, CH), 110.9 (CH), 109.3 (CH), 70.3 (d, JP–C = 5.4 Hz), 55.9 (CH3), 55.8 (CH3), 24.1 (d, JP–C = 4.2 Hz, CH3), 24.0 (d, JP–C = 4.2 Hz, CH3). 31P NMR (CDCl3) δ: 18.0. MS (ESI) m/z: 329.2 [M + H]+ (calcd for C16H26O5P+ 329.1).O), 1105 (P–O). 1H NMR (CDCl3) δ: 7.37 (dd, J = 22.4 Hz, J = 17.4 Hz, 1H), 7.00 (d, J = 1.1 Hz, 1H), 6.96 (d, J = 9.3 Hz, 1H), 6.79 (d, J = 8.0 Hz, 1H), 6.07 (t, J = 17.2 Hz, 1H), 5.99 (s, 2H), 4.74–4.65 (m, 2H), 1.36 (d, J = 6.2 Hz, 6H), 1.31 (d, J = 6.2 Hz, 6H). 13C NMR (CDCl3) δ: 149.3, 148.2, 147.3 (d, JP–C = 7.2 Hz, CH), 129.4 (d, JP–C = 23.8 Hz), 123.7 (CH), 113.0 (d, JP–C = 192.1 Hz, CH), 107.2 (d, JP–C = 225.5 Hz, CH), 101.4 (CH), 70.3 (d, JP–C = 5.5 Hz), 24.0 (d, JP–C = 4.0 Hz, CH3), 23.9 (d, JP–C = 4.2 Hz, CH3). 31P NMR (CDCl3) δ: 17.8. MS (ESI) m/z: 313.3 [M + H]+ (calcd for C15H22O5P+ 313.1).O), 1105 (P–O), 754 (C–Cl). 1H NMR (CDCl3) δ: 7.83 (dd, J = 22.6 Hz, J = 17.5 Hz, 1H), 7.60–7.57 (m, 1H), 7.40–7.38 (m, 1H), 7.31–7.27 (m, 2H), 6.31 (t, J = 17.6 Hz, 1H), 4.79–4.70 (m, 2H), 1.36 (dd, J = 12.7 Hz, J = 6.2 Hz, 12H). 13C NMR (CDCl3) δ: 143.0 (d, JP–C = 7.8 Hz), 134.4, 133.2 (d, JP–C = 23.7 Hz), 130.8, 130.0 (d, JP–C = 1.0 Hz), 127.3, 127.0, 119.8, 117.9, 70.7 (d, JP–C = 5.7 Hz), 24.1 (d, JP–C = 4.0 Hz), 24.0 (d, JP–C = 4.8 Hz). 31P NMR (CDCl3) δ: 16.0. HR MS (ESI) m/z: 303.0914 [M + H]+ (calcd for C14H21ClO3P+ 303.0911).O), 1153 (P–O), 756 (C–Cl). 1H NMR (CDCl3) δ: 7.51 (dd, J = 23.6 Hz, J = 17.8 Hz, 1H), 7.34 (d, J = 8.0 Hz, 2H), 7.18 (t, J = 8.0 Hz, 1H), 6.43 (t, J = 18.0 Hz, 1H), 4.80–4.72 (m, 2H), 1.38 (d, J = 6.2 Hz, 6H), 1.31 (d, J = 6.2 Hz, 6H). 13C NMR (CDCl3) δ: 141.3 (d, JP–C = 7.8 Hz), 134.5 (d, JP–C = 1.5 Hz), 132.8 (d, JP–C = 23.4 Hz), 129.6, 128.7, 125.8 (d, JP–C = 183.2 Hz), 70.5 (d, JP–C = 5.4 Hz), 24.1 (d, JP–C = 4.0 Hz), 23.9 (d, JP–C = 4.5 Hz). 31P NMR (CDCl3) δ: 14.6. HR MS (ESI) m/z: 337.0518 [M + H]+ (calcd for C14H20Cl2O3P+ 337.0522).O), 1108 (P–O). 1H NMR (CDCl3) δ: 7.45–7.34 (m, 3H), 7.02 (d, J = 8.6 Hz, 2H), 6.14 (t, J = 17.2 Hz, 1H), 4.71–4.63 (m, 2H), 1.32 (d, J = 6.2 Hz, 6H), 1.28 (d, J = 6.2 Hz, 6H). 13C NMR (CDCl3) δ: 163.7 (d, JF–C = 249.1 Hz), 146.4 (d, JP–C = 6.9 Hz, CH), 131.2 (dd, JF–C = 3.4 Hz, JP–C = 23.5 Hz), 129.4 (d, JP–C = 8.4 Hz, CH), 115.8 (d, JP–C = 21.8 Hz, CH), 115.3 (dd, JF–C = 2.0 Hz, JP–C = 191.7 Hz, CH), 70.5 (d, JP–C = 5.5 Hz, CH), 24.0 (d, JP–C = 4.0 Hz, CH3), 23.9 (d, JP–C = 4.0 Hz, CH3). 31P NMR (CDCl3) δ: 17.0. 19F NMR (CDCl3) δ: −110.2. MS (ESI) m/z: 287.3 [M + H]+ (calcd for C14H21FO3P+ 287.1).O), 1116 (P–O). 1H NMR (CDCl3) δ: 7.51–7.39 (m, 3H), 7.34 (d, J = 8.4 Hz, 2H), 6.24 (t, J = 17.2 Hz, 1H), 4.76–4.66 (m, 2H), 1.35 (d, J = 6.2 Hz, 6H), 1.30 (d, J = 6.2 Hz, 6H). 13C NMR (CDCl3) δ: 146.4 (d, JP–C = 7.0 Hz), 133.9 (d, JP–C = 23.5 Hz), 132.0, 129.0, 124.2, 116.2 (d, JP–C = 191.5 Hz), 70.5 (d, JP–C = 5.8 Hz), 24.0 (d, JP–C = 4.0 Hz), 23.9 (d, JP–C = 4.5 Hz). 31P NMR (CDCl3) δ: 16.6. MS (ESI) m/z: 347.2 [M + H]+ (calcd for C14H21BrO3P+ 347.0).O), 1109 (P–O). 1H NMR (CDCl3) δ: 7.70 (d, J = 8.4 Hz, 2H), 7.37 (dd, J = 17.6 Hz, J = 22.6 Hz, 1H), 7.20 (d, J = 8.4 Hz, 2H), 6.25 (t, J = 17.2 Hz, 1H), 4.73–4.65 (m, 2H), 1.35 (d, J = 6.2 Hz, 6H), 1.30 (d, J = 6.2 Hz, 6H). 13C NMR (CDCl3) δ: 146.4 (d, JP–C = 6.7 Hz), 138.0, 134.5 (d, JP–C = 23.3 Hz), 129.1, 127.6, 116.6 (d, JP–C = 191.0 Hz), 70.6 (d, JP–C = 5.7 Hz), 24.1 (d, JP–C = 4.0 Hz), 24.0 (d, JP–C = 4.5 Hz). 31P NMR (CDCl3) δ: 16.6. HR MS (ESI) m/z: 395.0270 [M + H]+ (calcd for C14H21IO3P+ 395.0268).O), 1103 (P–O). 1H NMR (CDCl3) δ: 7.49 (dd, J = 22.6 Hz, J = 17.4 Hz, 1H), 7.28 (d, J = 5.0 Hz, 1H), 7.13 (d, J = 3.3 Hz, 1H), 6.97 (t, J = 4.6 Hz, 1H), 5.95 (t, J = 16.8 Hz, 1H), 4.69–4.59 (m, 2H), 1.30 (d, J = 6.2 Hz, 6H), 1.26 (d, J = 6.2 Hz, 6H). 13C NMR (CDCl3) δ: 140.5 (d, JP–C = 26.7 Hz), 140.2 (d, JP–C = 7.6 Hz, CH), 129.9 (CH), 127.9 (d, JP–C = 1.1 Hz, CH), 127.8, 114.1 (d, JP–C = 193.1 Hz, CH), 70.5 (d, JP–C = 5.5 Hz, CH), 24.0 (d, JP–C = 4.0 Hz, CH3), 23.9 (d, JP–C = 4.0 Hz, CH3). 31P NMR (CDCl3) δ: 16.7. MS (ESI) m/z: 275.2 [M + H]+ (calcd for C12H20O3PS+ 275.0).O), 1132 (P–O). 1H NMR (CDCl3) δ: 7.47 (dd, J = 24.8 Hz, J = 1.4 Hz, 1H), 7.38 (d, J = 4.4 Hz, 4H), 7.34–7.29 (m, 1H), 4.71–4.68 (m, 2H), 2.06 (dd, J = 15.2 Hz, J = 1.4 Hz, 1H), 1.38 (d, J = 6.2 Hz, 6H), 1.31 (d, J = 6.2 Hz, 6H). 13C NMR (CDCl3) δ: 141.9 (d, JP–C = 12.5 Hz, CH), 135.9 (d, JP–C = 23.4 Hz), 129.4 (CH), 128.4 (CH), 128.2 (CH), 126.4, 70.3 (d, JP–C = 6.4 Hz, CH), 24.1 (d, JP–C = 4.2 Hz, CH3), 23.8 (d, JP–C = 4.5 Hz, CH3), 14.4 (d, JP–C = 9.7 Hz, CH3). 31P NMR (CDCl3) δ: 19.7. HR MS (ESI) m/z: 283.1456 [M + H]+ (calcd for C15H24O3P+ 283.1458).O), 1053 (P–O). 1H NMR (CDCl3) δ: 7.58–7.48 (m, 3H), 7.40–7.37 (m, 3H), 6.23 (t, J = 17.7 Hz, 1H), 3.78 (d, J = 11.1 Hz, 6H). 13C NMR (CDCl3) δ: 149.6 (d, JP–C = 6.6 Hz), 134.6 (d, JP–C = 23.1 Hz), 130.4, 128.9, 127.7, 112.3 (d, JP–C = 191.2 Hz), 52.4 (d, JP–C = 5.6 Hz). 31P NMR (CDCl3) δ: 22.4. MS (ESI) m/z: 213.1 [M + H]+ (calcd for C10H14O3P+ 213.0).O), 1134 (P–O). 1H NMR (CDCl3) δ: 7.56–7.46 (m, 3H), 7.40–7.37 (m, 3H), 6.26 (t, J = 17.6 Hz, 1H), 4.17–4.09 (m, 4H), 1.36 (t, J = 7.0 Hz, 6H). 13C NMR (CDCl3) δ: 148.7 (d, JP–C = 6.7 Hz), 134.8 (d, JP–C = 23.0 Hz), 130.2, 128.8, 127.7, 113.9 (d, JP–C = 190.3 Hz), 61.8 (d, JP–C = 5.5 Hz), 16.4 (d, JP–C = 6.4 Hz). 31P NMR (CDCl3) δ: 19.5. MS (ESI) m/z: 241.3 [M + H]+ (calcd for C12H18O3P+ 241.1).O), 1065 (P–O). 1H NMR (CDCl3) δ: 7.56–7.46 (m, 3H), 7.39–7.37 (m, 3H), 6.26 (t, J = 17.6 Hz, 1H), 4.05–3.99 (m, 4H), 1.76–1.68 (m, 4H), 0.97 (d, J = 7.4 Hz, 6H). 13C NMR (CDCl3) δ: 148.8 (d, JP–C = 6.7 Hz), 134.8 (d, JP–C = 23.2 Hz), 130.3, 128.9, 127.7, 113.6 (d, JP–C = 191.0 Hz), 67.4 (d, JP–C = 5.8 Hz), 23.8 (d, JP–C = 6.5 Hz), 10.0. 31P NMR (CDCl3) δ: 19.6. MS (ESI) m/z: 269.2 [M + H]+ (calcd for C14H22O3P+ 269.1).O), 1068 (P–O). 1H NMR (CDCl3) δ: 7.52–7.42 (m, 3H), 7.36–7.33 (m, 3H), 6.24 (t, J = 17.6 Hz, 1H), 4.06–3.98 (m, 4H), 1.67–1.60 (m, 4H), 1.43–1.35 (m, 4H), 0.89 (t, J = 7.4 Hz, 6H). 13C NMR (CDCl3) δ: 148.7 (d, JP–C = 6.6 Hz, CH), 134.8 (d, JP–C = 23.1 Hz), 130.2 (CH), 128.8 (CH), 127.7 (CH), 113.8 (d, JP–C = 190.4 Hz, CH), 65.5 (d, JP–C = 5.7 Hz, CH2), 32.5 (d, JP–C = 6.3 Hz, CH2), 18.7 (CH2), 13.6 (CH3). 31P NMR (CDCl3) δ: 19.6. MS (ESI) m/z: 297.3 [M + H]+ (calcd for C16H26O3P+ 297.1).O), 1047 (P–O). 1H NMR (CDCl3) δ: 7.57–7.47 (m, 3H), 7.39–7.37 (m, 3H), 6.26 (t, J = 17.6 Hz, 1H), 3.85–3.80 (m, 4H), 2.00–1.93 (m, 2H), 0.95 (d, J = 6.8 Hz, 9H). 13C NMR (CDCl3) δ: 148.9 (d, JP–C = 6.5 Hz), 134.8 (d, JP–C = 23.3 Hz), 130.3, 128.9, 127.7, 113.4 (d, JP–C = 191.7 Hz), 71.9 (d, JP–C = 6.1 Hz), 29.1 (d, JP–C = 6.7 Hz), 18.7 (d, JP–C = 1.9 Hz). 31P NMR (CDCl3) δ: 19.4. HR MS (ESI) m/z: 297.1618 [M + H]+ (calcd for C16H26O3P+ 297.1614).O), 1120 (P–O). 1H NMR (CDCl3) δ: 7.77–7.72 (m, 4H), 7.55–7.44 (m, 9H), 7.36–7.35 (m, 3H), 6.83 (dd, J = 22.4 Hz, J = 17.4 Hz, 1H). 13C NMR (CDCl3) δ: 147.6 (d, JP–C = 3.5 Hz, CH), 135.1 (d, JP–C = 17.8 Hz), 132.8 (d, JP–C = 105.5 Hz), 131.9 (d, JP–C = 2.5 Hz, CH), 131.4 (d, JP–C = 10.0 Hz, CH), 130.1, 128.8 (d, JP–C = 27.8 Hz, CH), 128.7 (CH), 127.8 (CH), 119.0 (d, JP–C = 103.9 Hz, CH). 31P NMR (CDCl3) δ: 24.8. MS (ESI) m/z: 305.0 [M + H]+ (calcd for C20H18OP+ 305.1).O), 1026 (P–O). 1H NMR (CDCl3) δ: 7.47 (dd, J = 22.6 Hz, J = 17.5 Hz, 1H), 7.39 (d, J = 8.1 Hz, 2H), 7.18 (d, J = 8.1 Hz, 2H), 6.19 (t, J = 17.7 Hz, 1H), 4.16–4.08 (m, 4H), 2.37 (s, 3H), 1.35 (t, J = 7.0 Hz, 6H). 13C NMR (CDCl3) δ: 148.8 (d, JP–C = 6.6 Hz), 140.6, 132.1 (d, JP–C = 23.2 Hz), 129.5, 127.7, 112.4 (d, JP–C = 190.8 Hz), 61.8 (d, JP–C = 5.4 Hz), 21.4, 16.4 (d, JP–C = 6.5 Hz). 31P NMR (CDCl3) δ: 19.9. MS (ESI) m/z: 255.0 [M + H]+ (calcd for C13H20O3P+ 255.1).O), 1028 (P–O). 1H NMR (CDCl3) δ: 7.46–7.36 (m, 3H), 6.86 (d, J = 8.8 Hz, 2H), 6.05 (t, J = 17.6 Hz, 1H), 4.12–4.04 (m, 4H), 3.79 (s, 3H), 1.31 (t, J = 7.1 Hz, 6H). 13C NMR (CDCl3) δ: 161.2, 148.4 (d, JP–C = 6.9 Hz, CH), 129.3 (CH), 127.6 (d, JP–C = 23.7 Hz), 114.2 (CH), 110.6 (d, JP–C = 191.7 Hz, CH), 61.7 (d, JP–C = 5.3 Hz, CH2), 55.3 (CH3), 16.4 (d, JP–C = 6.5 Hz, CH3). 31P NMR (CDCl3) δ: 20.4. MS (ESI) m/z: 271.3 [M + H]+ (calcd for C13H20O4P+ 271.1).O), 1075 (P–O), 806 (C–Cl). 1H NMR (CDCl3) δ: 7.86 (dd, J = 22.6 Hz, J = 17.5 Hz, 1H), 7.61–7.58 (m, 1H), 7.41–7.39 (m, 1H), 7.31–7.28 (m, 2H), 6.30 (t, J = 17.7 Hz, 1H), 4.20–4.12 (m, 4H), 1.37 (t, J = 7.0 Hz, 6H). 13C NMR (CDCl3) δ: 144.0 (d, JP–C = 7.8 Hz), 134.5, 133.2 (d, JP–C = 23.5 Hz), 131.0, 130.0, 127.4, 127.0, 117.2 (d, JP–C = 33.0 Hz), 62.1 (d, JP–C = 5.6 Hz), 16.4 (d, JP–C = 6.4 Hz). 31P NMR (CDCl3) δ: 18.2. MS (ESI) m/z: 275.2 [M + H]+ (calcd for C12H17ClO3P+ 275.0).O), 1151 (P–O), 764 (C–Cl). 1H NMR (CDCl3) δ: 7.53 (dd, J = 23.7 Hz, J = 17.9 Hz, 1H), 7.36 (d, J = 7.7 Hz, 2H), 7.19 (t, J = 8.2 Hz, 1H), 6.42 (t, J = 18.2 Hz, 1H), 4.21–4.14 (m, 4H), 1.38 (d, J = 7.0 Hz, 6H). 13C NMR (CDCl3) δ: 142.1 (d, JP–C = 7.6 Hz), 134.5 (d, JP–C = 1.4 Hz), 129.8, 128.7, 127.9, 124.2 (d, JP–C = 182.5 Hz), 62.2 (d, JP–C = 5.4 Hz), 16.4 (d, JP–C = 6.4 Hz). 31P NMR (CDCl3) δ: 17.0. MS (ESI) m/z: 309.2 [M + H]+ (calcd for C12H16Cl2O3P+ 309.0).O), 1038 (P–O). 1H NMR (CDCl3) δ: 7.33 (dd, J = 22.4 Hz, J = 17.4 Hz, 1H), 6.94 (s, 1H), 6.90 (d, J = 8.0 Hz, 1H), 6.73 (d, J = 8.0 Hz, 1H), 5.98 (t, J = 17.4 Hz, 1H), 5.93 (s, 2H), 4.09–4.01 (m, 4H), 1.28 (t, J = 7.1 Hz, 6H). 13C NMR (CDCl3) δ: 149.4, 148.4, 148.3 (d, JP–C = 3.8 Hz, CH), 129.3 (d, JP–C = 23.8 Hz), 123.9 (CH), 112.3 (CH), 110.4 (CH), 108.4 (CH), 106.2 (CH), 101.5 (CH2), 61.7 (d, JP–C = 5.5 Hz, CH2), 16.3 (d, JP–C = 6.4 Hz, CH3). 31P NMR (CDCl3) δ: 19.6. MS (ESI) m/z: 285.2 [M + H]+ (calcd for C13H18O5P+ 285.0).O), 1106 (P–O). 1H NMR (CDCl3) δ: 8.18 (dd, J = 29.4 Hz, J = 47.7 Hz, 1H), 7.55 (d, J = 1.3 Hz, 1H), 7.30 (d, J = 8.2 Hz, 1H), 6.83 (d, J = 8.2 Hz, 1H), 6.83 (s, 2H), 4.22–4.14 (m, 4H), 4.13–4.05 (m, 4H), 1.37 (t, J = 7.0 Hz, 6H), 1.24 (t, J = 7.0 Hz, 6H). 13C NMR (CDCl3) δ: 161.1 (d, JP–C = 2.2 Hz), 150.1, 147.6, 128.3 (dd, JP–C = 22.6 Hz, JP–C = 8.8 Hz), 127.8, 110.6, 107.9, 101.6, 62.6 (d, JP–C = 5.2 Hz), 62.4 (d, JP–C = 5.8 Hz), 16.3 (d, JP–C = 6.6 Hz), 16.1 (d, JP–C = 6.6 Hz). 31P NMR (CDCl3) δ: 18.3 (d, JP–P = 49.5 Hz), 12.8 (d, JP–P = 49.5 Hz). HR MS (ESI) m/z: 421.1178 [M + H]+ (calcd for C17H27O8P2+ 421.1176).O), 1036 (P–O). 1H NMR (CDCl3) δ: 7.40 (dd, J = 22.4 Hz, J = 17.4 Hz, 1H), 7.01 (d, J = 1.3 Hz, 1H), 6.97 (d, J = 8.0 Hz, 1H), 6.80 (d, J = 8.0 Hz, 1H), 6.09–6.00 (m, 3H), 4.03–3.97 (m, 4H), 1.76–1.67 (m, 4H), 0.96 (t, J = 7.4 Hz, 6H). 13C NMR (CDCl3) δ: 149.4, 148.3 (d, JP–C = 5.0 Hz), 129.3 (d, JP–C = 23.7 Hz), 123.9, 112.3, 110.4, 108.4, 106.2, 101.5, 67.3 (d, JP–C = 5.5 Hz), 23.8 (d, JP–C = 6.5 Hz), 10.1. 31P NMR (CDCl3) δ: 20.1. HR MS (ESI) m/z: 313.1202 [M + H]+ (calcd for C15H22O5P+ 313.1199).O), 1036 (P–O). 1H NMR (CDCl3) δ: 7.38–7.29 (m, 11H), 6.90–6.87 (m, 2H), 6.76 (d, J = 7.9 Hz, 1H), 6.01 (t, J = 17.6 Hz, 1H), 5.96 (s, 2H), 5.07 (s, 2H), 5.05 (s, 2H). 13C NMR (CDCl3) δ: 149.6, 148.7 (d, JP–C = 7.2 Hz, CH), 136.3 (d, JP–C = 6.6 Hz), 129.2 (d, JP–C = 24.4 Hz), 128.6 (CH), 128.4 (CH), 127.9 (CH), 124.0 (CH), 111.0 (d, JP–C = 192.8 Hz, CH), 108.4 (CH), 106.2 (CH), 101.5 (CH2), 67.4 (d, JP–C = 5.2 Hz, CH2). 31P NMR (CDCl3) δ: 21.1. HR MS (ESI) m/z: 409.1196 [M + H]+ (calcd for C23H22O5P+ 409.1199).O), 1049 (P–O). 1H NMR (CDCl3) δ: 7.87 (dd, J = 23.0 Hz, J = 17.5 Hz, 1H), 7.47 (dd, J = 7.5 Hz, J = 1.6 Hz, 1H), 7.39–7.24 (m, 13H), 6.24 (t, J = 18.1 Hz, 1H), 5.11 (d, J = 2.8 Hz, 2H), 5.09 (d, J = 2.7 Hz, 2H). 13C NMR (CDCl3) δ: 144.4 (d, JP–C = 8.1 Hz, CH), 136.1 (d, JP–C = 6.5 Hz), 134.6, 133.0 (d, JP–C = 23.9 Hz), 131.0 (CH), 130.1 (CH), 128.6 (CH), 128.4 (CH), 128.0 (CH), 127.4 (d, JP–C = 1.0 Hz, CH), 127.0 (CH), 116.9 (d, JP–C = 190.9 Hz, CH), 67.5 (d, JP–C = 5.6 Hz, CH2). 31P NMR (CDCl3) δ: 19.2. HR MS (ESI) m/z: 399.0909 [M + H]+ (calcd for C22H21ClO3P+ 399.0911).O), 1159 (P–O). 1H NMR (CDCl3) δ: 7.37 (dd, J = 22.4 Hz, J = 17.4 Hz, 1H), 7.02 (dd, J = 8.2 Hz, J = 1.5 Hz, 1H), 6.98 (d, J = 1.5 Hz, 1H), 6.81 (d, J = 8.3 Hz, 1H), 6.04 (t, J = 17.4 Hz, 1H), 4.03–3.96 (m, 4H), 3.85 (s, 6H), 1.66–1.59 (m, 4H), 1.41–1.32 (m, 4H), 0.87 (t, J = 7.4 Hz, 6H). 13C NMR (CDCl3) δ: 150.9, 149.1, 148.5 (d, JP–C = 6.9 Hz, CH), 127.8 (d, JP–C = 23.5 Hz), 122.0 (CH), 111.0 (d, JP–C = 191.9 Hz, CH), 110.9 (CH), 109.3 (CH), 65.5 (CH3), 65.4 (CH3), 55.9 (d, JP–C = 8.9 Hz, CH2), 32.5 (d, JP–C = 6.5 Hz, CH2), 18.7 (CH2), 13.6 (CH3). 31P NMR (CDCl3) δ: 19.8. HR MS (ESI) m/z: 357.1823 [M + H]+ (calcd for C18H30O5P+ 357.1825).Acknowledgements
We gratefully acknowledge the National Natural Science Foundation of China (No. 21302042 and 21172055), Department of Henan Province Natural Science and Technology Foundation (No. 142102210410), Natural Science Foundation in Henan Province Department of Education (No. 14B150053 and 17A150005), the Program for Innovative Research Team from Zhengzhou (No. 131PCXTD605).Notes and references
- (a) M. Maffei, Curr. Org. Chem., 2004, 1, 355 CAS; (b) L. Coudray and J. L. Montchamp, Eur. J. Org. Chem., 2008, 3601 CrossRef CAS PubMed.
- (a) Z. C. Duan, X. P. Hu, D. Y. Wang, J. D. Huang, S. B. Yu, J. Deng and Z. Zheng, Adv. Synth. Catal., 2008, 10, 3141 Search PubMed; (b) C. Garzon, M. Attolini and M. Maffei, Eur. J. Org. Chem., 2013, 3653 CrossRef CAS; (c) J. Ito, K. Fujii and H. Nishiyama, Chem.–Eur. J., 2013, 19, 601 CrossRef CAS PubMed; (d) D. Ender, H. Wahl and K. Papadopoulos, Tetrahedron, 1997, 53, 13961 Search PubMed; (e) H. Inoue, H. Tsubouchi, Y. Nagaoka and K. Tomioka, Tetrahedron, 2002, 58, 83 CrossRef CAS.
- (a) S. Salzinger, B. S. Soller, A. Plikhta, U. B. Seemann, E. Herdtweck and B. Rieger, J. Am. Chem. Soc., 2013, 135, 13030 CrossRef CAS PubMed; (b) S. Jin and K. E. Gonsalves, Macromolecules, 1998, 31, 1010 CrossRef CAS; (c) J. A. Russell, F. A. Ching, H. I. Tashtoush, J. E. Russell and D. F. Dedolph, J. Org. Chem., 1991, 56, 663 CrossRef; (d) M. Ruiz, M. C. Fernández, S. Conde, A. Diaz and J. M. Quintela, Synlett, 1999, 1903 CrossRef CAS.
- (a) A. Quntar, O. Baum, R. Reich and M. Srebnik, Arch. Pharm., 2004, 337, 76 CrossRef PubMed; (b) Z. Liu, N. MacRitchie, S. Pyne, N. J. Pyne and R. Bittman, Bioorg. Med. Chem., 2013, 21, 2503 CrossRef CAS PubMed; (c) T. Minami and J. Motoyoshiya, Synthesis, 1992, 333 CrossRef CAS; (d) M. R. Harnden, A. Parkin, M. J. Parratt and R. M. Perkins, J. Med. Chem., 1993, 36, 1343 CrossRef CAS PubMed.
- (a) X. Mi, M. M. Huang, J. Y. Zhang, C. Y. Wang and Y. J. Wu, Org. Lett., 2013, 15, 6266 CrossRef CAS PubMed; (b) Y. M. Li, Y. Shen, K. J. Chang and S. D. Yang, Tetrahedron Lett., 2014, 55, 2119 CrossRef CAS; (c) J. W. Yuan, Y. Z. Li, L. R. Yang, W. P. Mai, P. Mao, Y. M. Xiao and L. B. Qu, Tetrahedron, 2015, 71, 8178 CrossRef CAS.
- (a) G. Evano, K. Tadiparthi and F. Couty, Chem. Commun., 2011, 47, 179 RSC; (b) L. L. Mao, A. X. Zhou, N. Liu and S. D. Yang, Synlett, 2014, 25, 2727 CrossRef CAS; (c) J. Hu, N. Zhao, B. Yang, G. Wang, L. N. Guo, Y. M. Liang and S. D. Yang, Chem.–Eur. J., 2011, 17, 5516 CrossRef CAS PubMed; (d) S. Thielges, P. Bisseret and J. Eustache, Org. Lett., 2005, 7, 681 CrossRef CAS PubMed.
- (a) L. Liu, Y. Lv, Y. L. Wu, X. Gao, Z. P. Zeng, Y. X. Gao, G. Tang and Y. F. Zhao, RSC Adv., 2014, 4, 2322 RSC; (b) Y. L. Wu, L. Liu, K. L. Yan, P. X. Xu, Y. X. Gao and Y. F. Zhao, J. Org. Chem., 2014, 79, 8118 CrossRef CAS PubMed.
- (a) J. F. Xue, S. F. Zhou, Y. Y. Liu, X. Q. Pan, J. P. Zou and O. T. Asekun, Org. Biomol. Chem., 2015, 13, 4896 RSC; (b) Q. W. Gui, L. Hu, X. Chen, J. D. Liu and Z. Tan, Chem. Commun., 2015, 51, 13922 RSC.
- (a) P. Zhong, X. Huang and Z. X. Xiong, Synlett, 1999, 6, 721 CrossRef; (b) P. Zhong, Z. X. Xiong and X. Huang, Synth. Commun., 2000, 30, 273 CrossRef CAS; (c) L. B. Han and M. Tanaka, J. Am. Chem. Soc., 1996, 118, 1571 CrossRef CAS; (d) L. B. Han, C. Q. Zhao and M. Tanaka, J. Org. Chem., 2001, 66, 5929 CrossRef CAS PubMed.
- (a) G. W. Kabalka and S. K. Guchhait, Org. Lett., 2003, 5, 729 CrossRef CAS PubMed; (b) G. Axelrad, S. Laosooksathit and R. Engel, Synth. Commun., 1980, 10, 933 CrossRef CAS.
- (a) G. W. Kabalka, S. K. Guchhait and A. Naravane, Tetrahedron Lett., 2004, 45, 4685 CrossRef CAS; (b) M. M. D. Pramanik, A. K. Chaturvedi and N. Rastogi, Chem. Commun., 2014, 50, 12896 RSC.
- (a) E. J. Corey and H. Estreicher, J. Am. Chem. Soc., 1978, 100, 6294 CrossRef CAS; (b) A. G. M. Barrett and G. G. Graboski, Chem. Rev., 1986, 86, 751 CrossRef CAS; (c) J. T. Liu, Y. J. Jang, Y. K. Shih, S. R. Hu, C. M. Chu and C. F. Yao, J. Org. Chem., 2001, 66, 6021 CrossRef CAS PubMed.
- (a) J. C. Anderson, A. S. Kalogirou and G. J. Tizzard, Tetrahedron, 2014, 70, 9337 CrossRef CAS; (b) C. F. Yao, C. M. Chu and J. T. Liu, J. Org. Chem., 1998, 63, 719 CrossRef CAS PubMed; (c) J. Y. Liu, J. T. Liu and C. F. Yao, Tetrahedron Lett., 2001, 42, 3613 CrossRef CAS.
- Y. J. Jang, M. C. Yan, Y. F. Lin and C. F. Yao, J. Org. Chem., 2004, 69, 3961 CrossRef CAS PubMed.
- (a) W. P. Mai, G. C. Sun, J. T. Wang, G. Song, P. Mao, L. R. Yang, J. W. Yuan, Y. M. Xiao and L. B. Qu, J. Org. Chem., 2014, 79, 8094 CrossRef CAS PubMed; (b) W. P. Mai, J. T. Wang, L. R. Yang, J. W. Yuan, Y. M. Xiao, P. Mao and L. B. Qu, Org. Lett., 2014, 16, 204 CrossRef CAS PubMed; (c) W. M. Zhao, X. L. Chen, J. W. Yuan, L. B. Qu, L. K. Duan and Y. F. Zhao, Chem. Commun., 2014, 50, 2018 RSC.
- (a) J. W. Yuan, Y. Z. Li, L. R. Yang, W. P. Mai, P. Mao, Y. M. Xiao and L. B. Qu, Tetrahedron, 2015, 71, 8178 CrossRef CAS; (b) J. W. Yuan, Y. Z. Li, W. P. Mai, L. R. Yang and L. B. Qu, Tetrahedron, 2016, 72, 3084 CrossRef CAS; (c) Y. Unoh, K. Hirano, T. Satoh and M. Miura, Angew. Chem., Int. Ed., 2013, 52, 12975 CrossRef CAS PubMed; (d) Y. R. Chen and W. L. Duan, J. Am. Chem. Soc., 2013, 135, 16754 CrossRef CAS PubMed.
- (a) X. L. Chen, X. Li, J. W. Yuan, L. B. Qu, S. H. Wang, H. Y. Shi, Y. C. Tang and L. K. Duan, Tetrahedron, 2013, 69, 4047 CrossRef CAS; (b) V. Kunzmann, E. Bauer and M. N. Wilhelm, N. Engl. J. Med., 1999, 340, 737 CrossRef CAS PubMed.
- (a) Y. M. Li, M. Sun, H. L. Wang, Q. P. Tian and S. D. Yang, Angew. Chem., Int. Ed., 2013, 52, 3972 CrossRef CAS PubMed; (b) J. Ke, Y. Tang, H. Yi, Y. Li, Y. Cheng, C. Liu and A. Lei, Angew. Chem., Int. Ed., 2015, 54, 6604 CrossRef CAS PubMed; (c) B. Zhang, C. G. Daniliuc and A. Studer, Org. Lett., 2014, 16, 250 CrossRef CAS PubMed; (d) X. Mi, C. Y. Wang, M. M. Huang, J. Y. Zhang, Y. S. Wu and Y. J. Wu, Org. Lett., 2014, 16, 3356 CrossRef CAS PubMed.
- X. Qi, S. H. Lee, J. Y. Kwon, Y. M. Kim, S. J. Kim, Y. S. Lee and J. Y. Yoon, J. Org. Chem., 2003, 68, 9140 CrossRef CAS PubMed.
- S. F. Zhou, D. P. Li, K. Liu, J. P. Zou and O. T. Asekun, J. Org. Chem., 2015, 80, 1214 CrossRef CAS PubMed.
- H. Krawczyk and Ł. Albrecht, Synthesis, 2005, 17, 2887 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Part of the experimental dedail and NMR spectra data. See DOI: 10.1039/c6ra19002b |
| This journal is © The Royal Society of Chemistry 2016 |