
Synthesis of magnetic microporous organic nanotube networks for adsorption application†
Minghong Zhou,
Hui Zhang,
Linfeng Xiong,
Zidong He,
Aiqing Zhong,
Tianqi Wang,
Yang Xu and
Kun Huang*
School of Chemistry and Molecular Engineering, East China Normal University, 500N, Dongchuan Road, Shanghai, 200241, P. R. China. E-mail: khuang@chem.ecnu.edu.cn
First published on 7th September 2016
Abstract
In this work, we report a novel synthesis of magnetic microporous organic nanotube networks (Fe3O4-MONNs) by an in situ hyper-cross-linking reaction between magnetic nanoparticles and core–shell bottlebrush copolymers. The resulting Fe3O4-MONNs magnetic hybrid materials display a hierarchically porous structure with nanotube morphology, large surface area (648 m2 g−1) and uniform mesochannels (∼4 nm). Due to abundant anionic carboxylate groups produced as end-groups after polylactide (PLA) core degradation in the bottlebrush copolymers, the Fe3O4-MONNs showed a selective adsorption behavior for the cationic dyes. Moreover, the Fe3O4-MONNs possess superparamagnetism and high saturation magnetization (19.8 emu g−1), which allows them to be easily separated by an external magnetic field and subsequently reused. Therefore, this work provides a promising method for the design and synthesis of magnetic microporous organic nanotube networks, which can be used for the practical separation of organic dyes, as well as having other potential applications in the fields of absorption, fast separation and heterogeneous catalysis.
1. Introduction
During the past decade, magnetic nanomaterials have been applied in many aspects such as catalysis, adsorption and separation because of their convenient manipulation, fast magnetic separation and multiple recycling.1–3 Especially, magnetic porous nanomaterials4 have attracted great interest due to their unique features such as a stable porous structure, large surface area, tunable pore size and magnetic properties. To date, a variety of synthetic methods combined with all kinds of porous materials such as metal organic frameworks (MOF),5,6 zeolites,7 carbon8,9 and mesoporous silica10–15 have been developed towards the fabrication of magnetic porous nanomaterials, including impregnation,16,17 the reverse-microemulsion method,11,18 self-assembly approach,19,20 surfactant-templated sol–gel techniques,14,15,21,22 covalent grafting,23 aerosol pyrolysis24 and etching approach.13,25 For example, highly dispersed magnetic mesoporous silica microspheres with well-defined core–shell structure, large and radially aligned mesopores were synthesized by Zhao and collaborators10 through a shearing assisted interface coassembly in biliquid phase systems. Although these strategies have achieved great success in preparing various magnetic porous materials, developing new porous materials and easy methods that construct more diverse architectures of magnetic porous structures and provide precise control over their pore structure and functionality as well as strong magnetic property still retains a scientific challenge.Among all kinds of porous materials, porous organic polymers (POPs) have attracted a particular attention due to their unique properties such as large surface area, low skeletal density and good chemical stability.26–28 In recent years, various new kinds of POPs have been developed such as polymers of intrinsic microporosity (PIMs),29,30 conjugated microporous polymers (CMPs),31–33 porous aromatic frameworks (PAFs),34 covalent organic frameworks (COFs)35–37 and hyper-cross-linked polymers (HCPs).38–42 All of these are aimed to meet the demand of porous organic materials with higher surface area and controlled pore sizes. However, the synthesis of magnetic porous polymeric hybrid materials has been relatively less explored. Very recently, some new kinds of porous polymeric networks have been used for coating magnetic particles, such as porous covalent triazine-based frameworks by a microwave-enhanced high-temperature ionothermal method,43 porous organic polymer nanoparticles by emulsion polymerization and Friedel–Crafts hyper-crosslinking reaction,44 and conjugated microporous polymer networks by Sonogashira coupling reaction.45 In our previous work, well-defined microporous organic nanotube networks can be prepared by the combination of molecular templating of core–shell bottlebrush copolymers and hyper-cross-linking via Friedel–Crafts (F–C) alkylation reaction.46,47 In addition, dopamine-terminated polystyrene (Dopa-PS) as the stable well-defined polymer ligands to anchor onto the iron oxide nanoparticle surfaces have been reported previously.48,49 These impressive results above provide us an inspiration for the fusion of microporous organic nanotube networks and magnetic nanoparticles through in situ hyper-cross-linking reaction.
Herein, a new class of magnetic microporous organic nanotube networks was successfully synthesized by combination of an in situ hyper-cross-linking reaction and molecule templating of core–shell bottlebrush copolymers. The proposed synthetic pathway is illustrated in Scheme 1. Firstly, core–shell bottlebrush copolymer precursors and magnetic nanoparticles Fe3O4@Dopa-PS coated by polymer ligands (Dopa-PS) were prepared by a grafting-from approach with the help of ring-opening and reversible addition–fragmentation chain transfer (RAFT) polymerizations as well as a ligand exchanged strategy, respectively. Subsequently, the outer polystyrene (PS) side chains from molecular bottlebrush copolymers and Fe3O4@Dopa-PS were then hyper-cross-linked to form a magnetic hybrid nanomaterial via a Friedel–Crafts alkylation reaction. Finally, by selective removal of the polylactide core retained in the bottlebrush copolymers, the microporous organic nanotube networks decorated by magnetic nanoparticles Fe3O4@Dopa-PS with a trimodal micro, meso- and macroporous architecture will be obtained. The micropore was obtained through the hyper-cross-linking reaction of PS shell layer, mesopore was generated by the selectively removed PLA core layer, while the meso/macropore resulted from a 3D continuous cross-linked network among bottlebrush copolymers and magnetic nanoparticles.
 | ||
| Scheme 1 Fabrication of magnetic microporous organic nanotube networks (Fe3O4-MONNs). | ||
2. Experiment section
2.1 Materials
Unless otherwise noted, all chemicals were used as received. Glycidyl methacrylate was distilled before use. 1,2-Dichloroethane (DCE) and N,N-dimethylformamide (DMF) were dried using CaH2 and distilled. Styrene was purified by passing over basic alumina. 2,2-Azobisisobutyronitrile (AIBN) and D,L-lactide were recrystallized from methanol and ethyl acetate, respectively. 2-Cyanoprop-2-yl-4-cyanodithilbenzoate (CPD)50 and S-1-dodecyl-S′-(α,α′-dimethyl-α′′-aceticacid)trithiocarbonate (TC)51 were synthesized according to literature procedures. Fe3O4-Cit magnetic nanoparticles were prepared by a solvothermal method according to literature procedures.522.2 Synthesis
(1) Preparation of dopamine-based chain transfer agent (Dopa-TC)49. Dicyclohexylcarbodiimide (DCC, 0.31 g, 1.5 mmol) and S-1-dodecyl-S′-(α,α′-dimethyl-α′′-acetic acid)thrithiocarbonate (TC, 0.36 g, 1.0 mmol) were dissolved in dry dichloromethane (5 mL), and then a suspension of N-hydroxysuccinimide (NHS, 0.17 g, 1.5 mmol) in dry dichloromethane (3 mL) was added at −10 °C under nitrogen atmosphere. The mixture was allowed to stir overnight at room temperature. The solvent was evaporated, and leaving a yellow crude product which was then subjected to column chromatography (SiO2: ethyl acetate/petroleum ether = 1/3). The NHS–TC with bright yellow solid in a quantitative yield was obtained.
The as attained NHS–TC (1.14 g, 2.47 mmol) and dopamine hydrochloride (0.56 g, 2.96 mmol) were stirred in absolute methanol (20 mL) with triethylamine (0.48 mL, 3.46 mmol) at room temperature under nitrogen atmosphere in the dark for 24 h. The solvent was evaporated, and leaving a yellow crude product which was then subjected to column chromatography (SiO2: ethyl acetate/petroleum ether = 1/2). The product (Dopa-TC) was obtained as a yellow solid in 60% yield.
(2) Synthesis of linear Dopa-PS polymer. Dopa-TC (60 mg), styrene (3 mL), AIBN (1.9 mg) and 1,4-dioxane (1.5 mL) were mixed in a Schlenk tube and degassed by 3 freeze–pump–thaw cycles. The RAFT polymerization was conducted at 70 °C for 8 h. Then, the polymerization was stopped by cooling to room temperature and opening the tube to air. The mixture was diluted with dichloromethane and precipitated in methanol for 3 times and dried under vacuum at room temperature for 24 h. Yield = 0.32 g.
GPC (PS stds): Mn = 3.3 × 103 g mol−1, Mw/Mn = 1.06. 1H NMR: n(PS) = 30.
(3) Preparation of Fe3O4@Dopa-PS by a ligand exchange reaction. Fe3O4-Cit magnetic particles (150 mg) and polymer Dopa-PS (100 mg) were dispersed in DMF (5 mL) and ultrasonically mixed for 3 h under nitrogen atmosphere. And then, the blown mixture was heated to 50 °C for 16 h. The sediment was separated with an external magnet. After it was washed by THF for several times, the Fe3O4@Dopa-PS magnetic particles were dried in vacuum.
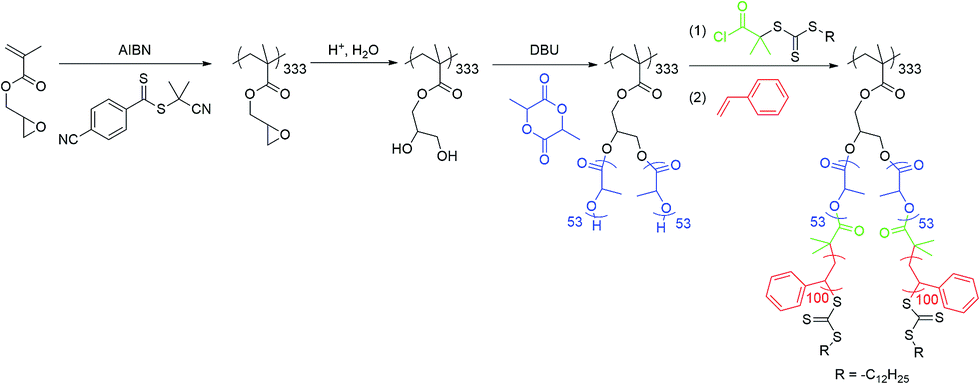
(1) Synthesis of PGM backbone and hydrolysis of epoxide groups. GM (2.3 mL), CPD (18 mg), AIBN (2.4 mg) and benzene (1.4 mL) were mixed in a Schlenk tube and degassed by 3 freeze–pump–thaw cycles. The polymerization was conducted at 60 °C for 12.5 h. Then, the reaction was stopped by cooling to room temperature and opening the tube to air. The mixture was diluted with dichloromethane and precipitated in methanol 3 times and dried under vacuum at 25 °C for 24 h. Yield = 1.95 g (81%). GPC (PS stds): Mn = 17.7 × 103 g mol−1, Mw/Mn = 1.22.
PGM (1.95 g), THF (40 mL) and acetic acid (80 mL) were mixed in a 500 mL round-bottom flask. The reaction mixture was stirred and placed in an oil bath at 60 °C, followed by the slow addition of 123 mL water over the course of 1 h. After stirring for 24 h at 60 °C, the solvent was removed on a rotary evaporator. The isolated polymer was precipitated from THF into diethyl ether 3 times and dried under vacuum at 25 °C for 24 h. Yield = 1.97 g (94%). 1H NMR: end-group conversion >95%.
(2) Synthesis of PGM-g-PLA. Hydrolyzed PGM (15 mg) and D,L-lactide (1.08 g) were added into a dried 50 mL round-bottom flask in a glovebox. Dried DMF (2.5 mL) was then added under nitrogen and the mixture was stirred until all polymer dissolved. 1,8-Diazbicyclo[5.4.0]undec-7-ene (DBU, 32.4 μL) was then injected into the flask. After stirring at room temperature for 2 h, the reaction was quenched by adding 165 mg of benzoic acid. The resulting polymer was precipitated from THF into methanol/water (1
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) 3 times and dried under vacuum at 25 °C for 24 h. Yield = 0.75 g (68.5%). GPC (PS stds): Mn = 3.6 × 105 g mol−1, Mw/Mn = 1.14. 1H NMR: n(PLA) = 53.
:1) 3 times and dried under vacuum at 25 °C for 24 h. Yield = 0.75 g (68.5%). GPC (PS stds): Mn = 3.6 × 105 g mol−1, Mw/Mn = 1.14. 1H NMR: n(PLA) = 53.
(3) Synthesis of PGM-g-PLA–TC. Oxalyl chloride (0.364 mL, 4.3 × 10−3 mol) and TC (0.20 g, 4.3 × 10−4 mol) were mixed in dry CH2Cl2 (5 mL) under nitrogen atmosphere and stirred at room temperature until gas evolution stopped (∼2 h). Excess reagents were then removed under vacuum, and the residue was redissolved in dry CH2Cl2 (10 mL), followed by the addition of poly(GM-g-LA)–OH (0.72 g in 10 mL of CH2Cl2). The reaction was allowed to proceed for 24 hours at room temperature, after which the contents were precipitated in methanol. The polymer was then redissolved in CH2Cl2, precipitated in methanol for twice, and dried in a vacuum oven overnight. Yield = 0.62 g (79.4%). GPC (PS stds): Mn = 3.8 × 105 g mol−1, Mw/Mn = 1.15. 1H NMR: end-group conversion >95%.
(4) Synthesis of PGM-g-(PLA-b-PS) bottlebrush copolymer. The modified polymer (100 mg) was then mixed with AIBN (0.18 mg), St (2.5 mL) and 1,4-dioxane (1.5 mL) in a reaction vessel and degassed by 3 freeze–pump–thaw cycles. The reaction was then conducted at 50 °C for 24 h. The reaction was stopped by cooling to room temperature and opening the flask to air. The resulting reaction mixture was then precipitated from THF into methanol 3 times and dried under vacuum at 25 °C for 24 h. Yield = 0.28 g (8%). GPC (PS stds): Mn = 7.9 × 105 g mol−1, Mw/Mn = 1.22. 1H NMR: n(St) = 100.
2.3 Dye adsorption experiments
Two anionic dyes and four cationic dyes were chosen for the adsorption experiments. Their structures are shown in Fig. 4. All the dye's concentrations were measured from the dyes' calibration curves. The corresponding UV-Vis spectra of dyes were recorded with a UV-2400 spectrophotometer.Saturated adsorption experiments were conducted with an initial dye concentration of 0.5 mg mL−1, except for the calcein dye which concentration was 0.04 mg mL−1. All of the volume of the dyes was 20 mL. 5 mg magnetic Fe3O4-MONNs adsorbents were immersed in the prepared dye solution for one day to reach their adsorption equilibrium. The equilibrium adsorption capacity (Qeq) of the six dyes was defined as follows:

2.4 Desorption and reusability experiments
Safranine T (ST) dye was used as an example for desorption and reusability study in this experiment. Typically, 10 mg of Fe3O4-MONNs adsorbents were immersed in 20 mL of safranine T (ST) solution (2 mg mL−1) for 8 h. The ST-adsorbed Fe3O4-MONNs were separated by magnetic field. Then, the ST-adsorbed Fe3O4-MONNs were washed by deionized water for three times and dried under vacuum. The supernatant solution was analyzed by UV-Vis spectra. After that, 20 mL of acetic acid/methanol (3% v/v) was added to the ST-loaded Fe3O4-MONNs for 12 h. The eluent solution was analyzed by UV-Vis spectra. The Fe3O4-MONNs were collected by magnetic separation and washed by methanol (without acetic acid) until no ST was detected in the final elution and dried under vacuum. The regenerated Fe3O4-MONNs were then reused for next cycle of adsorption and desorption study, as described above. The recycles of adsorption experiments were conducted six times. The dye desorption percentage was calculated as the following:where C0 is the initial concentration of ST solution (mg mL−1), Ceq,ads and Ceq,des are the concentrations of ST solution at equilibrium in adsorption phase and desorption phase respectively (mg mL−1); Vads and Vdes are the volumes of ST solution and eluent solution (mL).
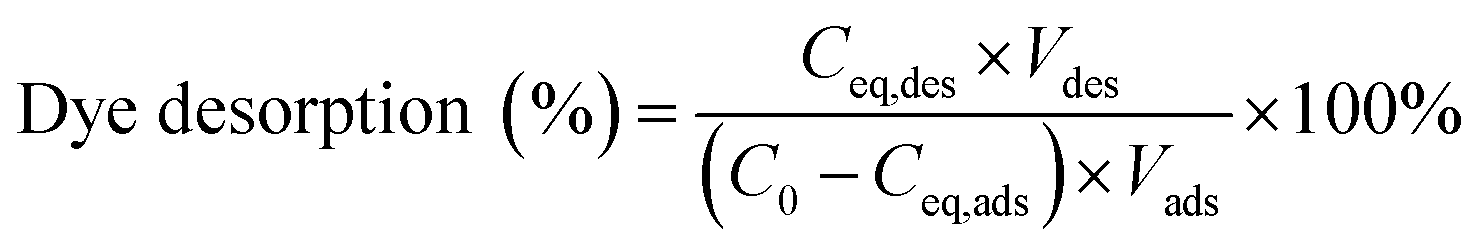
3. Results and discussion
3.1 Synthesis of magnetic nanoparticles (Fe3O4@Dopa-PS) and core–shell bottlebrush copolymer precursors
Inspired by the pioneering work by Woisel et al.,48 which uses dopamine-terminated polystyrene as the polymer ligands to anchor onto the iron oxide nanoparticle surfaces. Herein, a well-defined polymer (Dopa-PS30) with dopamine end group was synthesized by the RAFT polymerization as shown in the Fig. S1.† The 1H NMR, 13C NMR and mass spectrum for dopamine-based chain transfer agent (Dopa-TC) are provided in the ESI (Fig. S2–S5†). The structure of synthesized Dopa-PS was elucidated by 1H NMR. As shown in Fig. S6,† at 5.5 and 5.25 ppm for the two catechol hydroxyl groups, the peak at 3.26 ppm represent for the methylene protons linked to the trithiocarbonate. The signals at 3.06 and 2.42 ppm identified the methylene of dopamine. By taking into account the integration of the proton, the PS repeat units could be calculated to be 30. GPC trace of the Dopa-PS exhibited monomodal molecular weight distributions (Mw/Mn = 1.06) (Fig. S7, ESI†). Then, according to the previous reported method,48,49 Fe3O4@Dopa-PS30 magnetic nanoparticles were prepared by a ligand exchanged reaction based on the Fe3O4-Cit magnetic nanoparticles in DMF. The successful synthesis of Fe3O4@Dopa-PS was further certified by the peaks of the saturated C–H stretching vibration at 2853, 2924 cm−1 and aromatic stretching vibration at 3026 cm−1 together with the typical Fe–O stretching peaks at 594 and 431 cm−1 in the FTIR spectra (Fig. S8(b), ESI†). The well-defined Fe3O4@Dopa-PS NPs with a mean diameter of about 200 nm and fine spherical shape were also observed (see TEM image of Fig. S9, ESI†).Scheme 2 presents the synthesis of well-defined core–shell bottlebrush copolymers, which as previously described from a poly(glycidyl methacrylate) (PGM) backbone with an average degree of polymerization of 333 and a polydispersity index (PDI) of 1.22.53–55 From the 1H NMR spectrum of PGM-g-(PLA-b-PS) (Fig. S10, ESI†), every branch was composed of a PLA block with an average of 53 repeat units and a PS block with an average of 100 units. The molecular weight distributions of the core–shell bottlebrush copolymer precursor were characterized by GPC (Fig. S11, ESI†). GPC traces of all polymers exhibited low polydispersities (Mw/Mn < 1.22), which indicated efficient reinitiation and the formation of well-defined copolymer precursors.
 | ||
| Scheme 2 Synthesis procedure of core–shell bottlebrush copolymer precursors. | ||
3.2 Synthesis of magnetic microporous organic nanotube networks (Fe3O4-MONNs)
Hyper-cross-linking via Friedel–Crafts alkylation among aromatic rings has recently been used to produce HCPs and provide a rigid network. As illustrated in Scheme 1, the above PGM-g-(PLA-b-PS) core–shell bottlebrush copolymers and Fe3O4@Dopa-PS30 magnetic nanoparticles were then used as building blocks to prepare the magnetic microporous organic nanotube networks (Fe3O4-MONNs) through an in situ hyper-cross-linking reaction. In this study, the pendant phenyl groups from PS shell of bottlebrush copolymers and PS ligands of Fe3O4@Dopa-PS30 were used as cross-linkable components, formaldehyde dimethyl acetal as cross-linker and anhydrous FeCl3 as the catalyst. Subsequently, the as-prepared magnetic hybrid nanomaterials Fe3O4-MONNs were treated in basic condition to etch out PLA chains. The complete removal of PLA was confirmed by the disappearance of the characteristic PLA carbonyl stretch peak (1760 cm−1) in the FTIR spectrum (Fig. S8, ESI†). Furthermore, the aromatic ring skeleton vibrations peaks and the peaks of the Fe–O stretching still retained.Thermogravimetric analysis (TGA) was used to study the components of the Fe3O4-MONNs, and the results are shown in Fig. S12,† the trace amount of weight loss within the range of 100–200 °C is caused by the trace amount of water vapour adsorped by mesopore channels. The curve of Fe3O4-Cit (a) shows that the proportion of the stabilizing ligand citrate in magnetite particles is about 8.1 wt%, which is well coincident with literature value.52 From curve (b), we also find that the polymer layer of Fe3O4@Dopa-PS began to thermally decompose at about 250 °C, accompanied by a significant mass loss. The mass loss continued up to about 450 °C, after that, the mass kept relatively constant. The total mass loss of Fe3O4@Dopa-PS reaches to 16.1 wt% until 800 °C. The results revealed that the grafting of polymer Dopa-PS on the surface of Fe3O4-Cit by ligand exchanging was successful and effective. Due to forming hyper-cross-linking network, the initial decomposition temperature is much higher than that of Fe3O4@Dopa-PS nanoparticles and the total mass loss of Fe3O4-MONNs largely increases to 35.1 wt%.
To confirm the crystal structure of as-synthesized Fe3O4@Dopa-PS and Fe3O4-MONNs, XRD patterns of aforementioned magnetic nanoparticles were characterized. As show in Fig. 1A, the characteristic diffraction peaks of Fe3O4 are present in two samples, which can be assigned to the (220), (311), (400), (511) and (440) planes of a pure cubic Fe3O4 phase (JCPDS# 88-0315). The XRD patterns showed that Fe3O4@Dopa-PS and Fe3O4-MONNs have the crystalline nature of cubic Fe3O4, which agreed well with that of the standard magnetite. The above results suggest that ligand exchanging and in situ hyper-cross-linking did not affect the crystal structure and grain size of Fe3O4 phase, respectively.
 | ||
| Fig. 1 (A) X-ray diffraction (XRD) patterns of (a) Fe3O4@Dopa-PS, (b) Fe3O4-MONNs and comparison with the reference JCPDS no. 88-0315; (B) VSM magnetization curves of (a) Fe3O4@Dopa-PS and (b) Fe3O4-MONNs. | ||
Magnetic property is a crucial function of Fe3O4-MONNs for their applications in fast separation. The magnetic properties of Fe3O4@Dopa-PS and Fe3O4-MONNs were measured by a vibrating sample magnetometer (VSM). Fig. 1B shows the magnetization hysteresis loops of Fe3O4@Dopa-PS and Fe3O4-MONNs with an applied magnetic field sweeping from −15000 to 15000 Oe at room temperature. The saturation magnetization of the resulting Fe3O4@Dopa-PS can reach to 52.6 emu g−1. After the hyper-cross-linked with MONNs and PLA core removal, the saturation magnetization of the resultant Fe3O4-MONNs was reduced to 19.8 emu g−1, however, they were still superparamagnetic and strong enough for magnetic separation. When an external magnetic field was applied, the Fe3O4-MONNs could be attracted to the wall of vial within a few seconds.
The morphology of Fe3O4-MONNs was further confirmed by TEM. As shown in Fig. 2, it reveals the Fe3O4 nanoparticles were immobilized within the hyper-cross-linked microporous organic nanotube networks. The size and shape of the magnetic Fe3O4 core remained unchanged. And they were surrounded by the polymeric networks, which consisted of many nanotubes with average length about 50 nm and an average pore size about 4 nm in different orientations. Almost every cylindrical object around the magnetic nanoparticles appears to be hollow, confirming the formation of well-defined tubular structures. These results suggested that hyper cross-linking of the PS shell layer from Fe3O4@Dopa-PS and PGM-g-(PLA-b-PS) bottlebrush copolymers was important to produce the magnetic microporous organic nanotube networks, while the PLA core degradation ensured the formation of hollow tubular structure.
 | ||
| Fig. 2 TEM image of Fe3O4-MONNs. | ||
To confirm the existence of hierarchically porous structure, we investigated the N2 adsorption/desorption isotherms and pore size distributions of the resulting Fe3O4-MONNs. Fig. 3 shows a type IV curves with a distinct hysteresis loop in the high pressure region (P/P0 = 0.4–1.0), indicating that the Fe3O4-MONNs have mesoporous characteristics included capillary condensation that originate from the hyper-cross-linked network and the hollow cylindrical nanotubes units. While an adsorption uptake at low relative pressure confirmed the formation of micropore in the polymeric matrices.56 Because the hollow cylindrical nanotubes units and the hyper-cross-linked structure can provide internal surface area, the Fe3O4-MONNs was proved to have high specific surface area of 648 m2 g−1 (by Brunauer–Emmett–Teller analysis). The high surface area suggests that the hyper-cross-linked structure provided a more rigid network against to shrink or collapse of the pores during the isolation and drying processes. The total pore volume is 0.64 cm3 g−1. Permanent porosity is characteristic of rigid hyper-cross-linked networks, which can be confirmed by the pore size distribution curve (Fig. 3, inset). A trimodal distribution of pore sizes (average sizes at 0.6, 1.4 and 4.0 nm respectively, calculated by the nonlocal density functional theory (NLDFT) model) indicates the Fe3O4-MONNs possess hierarchically porous structure. Among them, the micropores exist in the wall of the nanotubes formed by the hyper-cross-linked shell layer; the mesopores ascribed to the PLA-templating of core–shell bottlebrush copolymers and the predominant close cross-linking aggregation of nanotube network units. The result of pores size coincides well with the TEM images.
 | ||
| Fig. 3 Nitrogen adsorption–desorption isotherms and pore size distribution of Fe3O4-MONNs based on NLDFT method. | ||
3.3 Study of dye adsorption in Fe3O4-MONNs
The resulting Fe3O4-MONNs possess a large surface area, good multi-porosity interconnectivity and sufficient magnetic property. Such novel type of Fe3O4-MONNs might lead to enhancing performances in heterogeneous catalysis, energy storage, adsorption, fast separation, drug delivery, etc. As Scheme 1 demonstrated, each nanotube unit of Fe3O4-MONNs has average about 666 carboxylic acid groups along the pore wall, which were produced as end-groups after PLA degradation. Therefore, the Fe3O4-MONNs with negatively charged pore walls can be expected to capture positively charged dyes. Herein, we studied their adsorption property toward different charged water-soluble organic dyes.The structure and abbreviation of the six hydrophilic dyes are listed in Fig. 4, and they can be divided into two types: the anionic dyes including Eosin B (EB), calcein (CA), and the cationic dyes including Methylene Blue (MB), Fuchsin Basic (FB), Rhodamine 6G (R6G), Safranine T (ST). The adsorption experiments were conducted in 3 mL aqueous solution, where the initial concentration of dyes was 0.01 mg mL−1. After addition of 4 mg Fe3O4-MONNs for several minutes, the color of MB, R6G, ST and FB solutions turned obviously colorless, while EB and CA solutions remained little changed (Fig. S13, ESI†). The saturated adsorption capacity (Qeq), which is an important parameter for an adsorbent in practical applications, was determined by UV-Vis spectra. As shown in Fig. 4, overall, the Qeqs of MB, FB, R6G, ST are 249, 259, 356 and 1105 mg g−1 respectively, which is much larger than that of the anionic dyes (EB for 10 mg g−1 and CA for 41 mg g−1). Taking the ST dye as an example, it showed that the red color of ST solution was almost disappeared just after shaking Fe3O4-MONNs with solution for several minutes. Once an external magnet was applied to the Fe3O4-MONNs nanomaterials, they will be attracted to the wall of vial within 10 seconds (Fig. 4, inset). The adsorption behavior is very fast due to existing of both micropores and mesopores in Fe3O4-MONNs, which can improve the diffusion and mass transport. After completely adsorption the dye, the initial brown color of Fe3O4-MONNs adsorbents turns into the adsorbed dye's color. In addition, the porous parameters of the ST-adsorbed Fe3O4-MONNs were also re-measured and the results showed that the BET specific surface area, microporous surface area, mesoporous surface area and total pore volume dramatically decreased (Table S1 and Fig. S14, ESI†), which demonstrated that the adsorbed ST dye has largely blocked the pore and channel within the Fe3O4-MONNs. The great difference of saturated adsorption capacity for cationic dyes and anionic dyes suggests the Fe3O4-MONNs could selectively adsorb the cationic dyes via the electrostatic interaction of anionic carboxylate groups on the pore wall. The above results show that the Fe3O4-MONNs possess the characterizations of high-performance selective absorption and fast magnetic separation!
 | ||
| Fig. 4 The structure and abbreviation of the six hydrophilic dyes and saturated adsorption capacities for EB, CA (anionic dyes) and FB, MB, R6G, ST (cationic dyes) at room temperature. The insets are photographs of (A) the solutions of ST after adsorption by the Fe3O4-MONNs for 3 min and then left under the magnet for 10 s and (B) initial solutions (0.01 mg mL−1 of ST). | ||
3.4 Desorption and reusability of Fe3O4-MONNs
The recycling ability of the adsorbent is crucial for its practical application. To evaluate the reusability of Fe3O4-MONNs, adsorption and desorption experiments were performed not only to separate and recover the dyes but also to restore the adsorption capacity of the exhausted adsorbent by elution with a magnetic separation step. Taking ST dye as an example, as shown in Fig. 5, the Fe3O4-MONNs still maintain high adsorption capacity of ST after six recycles, indicating the excellent recycling abilities of Fe3O4-MONNs for the separation of ST. Furthermore, the adsorbed ST can be desorbed from Fe3O4-MONNs by using acetic acid/methanol (3% v/v), and the desorption percentage still maintained above 95% after 6 runs. Interestingly, the re-generated porous Fe3O4-MONNs showed similar porous parameters as that of the as-synthesized porous Fe3O4-MONNs (Table S1 and Fig. S14, ESI†), which further proved the efficient adsorption/desorption ability and high stability of Fe3O4-MONNs. In addition, FTIR and TGA analysis also confirmed similar result. Once the adsorbed dye ST in Fe3O4-MONNs was desorbed, the characteristic stretch peaks from ST (1589 cm−1 and 1337 cm−1) in the FTIR spectra (Fig. S15, ESI†) completely disappeared and the total mass loss became higher (Fig. S16, ESI†). The effectiveness of the desorption might be attributed to the degree of dissociation of the –COOH along the pore wall suppressed in acidic methanol, which in turn reduced the electrostatic attraction between the cationic dyes of ST and the negatively charged pore wall of Fe3O4-MONNs. After elution, the Fe3O4-MONNs was magnetically retrieved and reused for the next cycle. All in all, the resulting Fe3O4-MONNs show wonderful adsorption and desorption properties towards organic dyes, which would have potential use to eliminate harmful organic dyes in the environment. | ||
| Fig. 5 Adsorption capacity and desorption percentage of ST in different recycles. | ||
4. Conclusions
In summary, we have developed a facile synthesis of magnetic microporous organic nanotube networks (Fe3O4-MONNs) by a combination of in situ hyper-cross-linking and molecule templating of core–shell bottlebrush copolymers. We had demonstrated the Fe3O4-MONNs could selectively adsorb the cationic dyes and achieve the fast separation by an external magnetic field. Compared with previously reported magnetic porous hybrid nanomaterials, the Fe3O4-MONNs have the following advantages: (1) with the high cross-linked density and robust covalent bond, Fe3O4-MONNs exhibit quite good chemical and thermal stability, which will further extent their applications even in harsh condition such as heterogeneous catalysis and bioseparation. (2) The Fe3O4-MONNs not only derive the microporous structure from hyper-cross-linked shell of the bottlebrush, but also maintain the mesopores tubular structure by the PLA-templating of core–shell bottlebrush copolymers. The hierarchically porous structure may largely improve their mass transport properties. (3) Unlike the porous carbon, a wide range of functionalities could be introduced to the Fe3O4-MONNs due to the synthetic diversification of the bottlebrush copolymers precursor. The functional groups can be introduced outside, inside or both outside and inside of the nanotubes. In this work, the nanotube interior of Fe3O4-MONNs were charged negative, which can selectively adsorb cationic dyes. (4) The Fe3O4-MONNs exhibit a high surface area, a high magnetization and the hierarchically porous structure. Furthermore, the magnetic properties of Fe3O4-MONNs could be adjusted by vary the ratio of Fe3O4@Dopa-PS NPs and bottlebrush copolymers. We believed that this novel hybrid material could be widely used in the field of absorption, fast bioseparation and heterogeneous catalysis.Acknowledgements
The work is supported by National Natural Science Foundation of China grant 51273066, 21574042, Shanghai Pujiang Program grant 13PJ1402300.Notes and references
- V. Polshettiwar, R. Luque, A. Fihri, H. Zhu, M. Bouhrara and J. M. Basset, Chem. Rev., 2011, 111, 3036–3075 CrossRef CAS PubMed.
- S. Shylesh, V. Schunemann and W. R. Thiel, Angew. Chem., Int. Ed., 2010, 49, 3428–3459 CrossRef CAS PubMed.
- D. Wang and D. Astruc, Chem. Rev., 2014, 114, 6949–6985 CrossRef CAS PubMed.
- J. Liu, S. Z. Qiao, Q. H. Hu and G. Q. Lu, Small, 2011, 7, 425–443 CrossRef CAS PubMed.
- J. Zheng, C. Cheng, W.-J. Fang, C. Chen, R.-W. Yan, H.-X. Huai and C.-C. Wang, CrystEngComm, 2014, 16, 3960 RSC.
- F. Ke, L.-G. Qiu, Y.-P. Yuan, X. Jiang and J.-F. Zhu, J. Mater. Chem., 2012, 22, 9497 RSC.
- Y. Deng, C. Deng, D. Qi, C. Liu, J. Liu, X. Zhang and D. Zhao, Adv. Mater., 2009, 21, 1377–1382 CrossRef CAS.
- Z. Wu, W. Li, P. A. Webley and D. Zhao, Adv. Mater., 2012, 24, 485–491 CrossRef CAS PubMed.
- J. E. Lee, S.-H. Yu, D. J. Lee, D.-C. Lee, S. I. Han, Y.-E. Sung and T. Hyeon, Energy Environ. Sci., 2012, 5, 9528 CAS.
- Q. Yue, J. Li, W. Luo, Y. Zhang, A. A. Elzatahry, X. Wang, C. Wang, W. Li, X. Cheng, A. Alghamdi, A. M. Abdullah, Y. Deng and D. Zhao, J. Am. Chem. Soc., 2015, 137, 13282–13289 CrossRef CAS PubMed.
- J. Kim, J. E. Lee, J. Lee, J. H. Yu, B. C. Kim, K. An, Y. Hwang, C.-H. Shin, J.-G. Park, J. Kim and T. Hyeon, J. Am. Chem. Soc., 2006, 128, 688–689 CrossRef CAS PubMed.
- J. Kim, H. S. Kim, N. Lee, T. Kim, H. Kim, T. Yu, I. C. Song, W. K. Moon and T. Hyeon, Angew. Chem., Int. Ed., 2008, 47, 8438–8441 CrossRef CAS PubMed.
- J. Ge, Q. Zhang, T. Zhang and Y. Yin, Angew. Chem., Int. Ed., 2008, 47, 8924–8928 CrossRef CAS PubMed.
- Y. Deng, D. Qi, C. Deng, X. Zhang and D. Zhao, J. Am. Chem. Soc., 2008, 130, 28–29 CrossRef CAS PubMed.
- Y. Deng, Y. Cai, Z. Sun, J. Liu, C. Liu, J. Wei, W. Li, C. Liu, Y. Wang and D. Zhao, J. Am. Chem. Soc., 2010, 132, 8466–8473 CrossRef CAS PubMed.
- H. H. P. Yiu, H. Niu, E. Biermans, G. van Tendeloo and M. J. Rosseinsky, Adv. Funct. Mater., 2010, 20, 1599–1609 CrossRef CAS.
- S. Alam, C. Anand, K. Ariga, T. Mori and A. Vinu, Angew. Chem., Int. Ed., 2009, 48, 7358–7361 CrossRef CAS PubMed.
- J. Kim, H. S. Kim, N. Lee, T. Kim, H. Kim, T. Yu, I. C. Song, W. K. Moon and T. Hyeon, Angew. Chem., Int. Ed., 2008, 47, 8438–8441 CrossRef CAS PubMed.
- C. B. W. Garcia, Y. Zhang, S. Mahajan, F. DiSalvo and U. Wiesner, J. Am. Chem. Soc., 2003, 125, 13310–13311 CrossRef CAS PubMed.
- C. Garcia, Y. Zhang, F. DiSalvo and U. Wiesner, Angew. Chem., Int. Ed., 2003, 42, 1526–1530 CrossRef CAS PubMed.
- W. Zhao, J. Gu, L. Zhang, H. Chen and J. Shi, J. Am. Chem. Soc., 2005, 127, 8916–8917 CrossRef CAS PubMed.
- P. Wang, Q. Shi, Y. Shi, K. K. Clark, G. D. Stucky and A. A. Keller, J. Am. Chem. Soc., 2009, 131, 182–188 CrossRef CAS PubMed.
- S. Giri, B. G. Trewyn, M. P. Stellmaker and V. S. Y. Lin, Angew. Chem., 2005, 117, 5166–5172 CrossRef.
- E. Ruiz-Hernández, A. López-Noriega, D. Arcos, I. Izquierdo-Barba, O. Terasaki and M. Vallet-Regí, Chem. Mater., 2007, 19, 3455–3463 CrossRef.
- Q. Zhang, J. Ge, J. Goebl, Y. Hu, Y. Sun and Y. Yin, Adv. Mater., 2010, 22, 1905–1909 CrossRef CAS PubMed.
- D. Wu, F. Xu, B. Sun, R. Fu, H. He and K. Matyjaszewski, Chem. Rev., 2012, 112, 3959–4015 CrossRef CAS PubMed.
- P. Kaur, J. T. Hupp and S. T. Nguyen, ACS Catal., 2011, 1, 819–835 CrossRef CAS.
- J.-X. Jiang and A. I. Cooper, in Functional Metal–Organic Frameworks: Gas Storage, Separation and Catalysis, ed. M. Schröder, Springer Berlin Heidelberg, Berlin, Heidelberg, 2010, pp. 1–33 Search PubMed.
- N. B. McKeown, B. Gahnem, K. J. Msayib, P. M. Budd, C. E. Tattershall, K. Mahmood, S. Tan, D. Book, H. W. Langmi and A. Walton, Angew. Chem., Int. Ed., 2006, 45, 1804–1807 CrossRef CAS PubMed.
- N. B. McKeown and P. M. Budd, Chem. Soc. Rev., 2006, 35, 675–683 RSC.
- J. Weber and A. Thomas, J. Am. Chem. Soc., 2008, 130, 6334–6335 CrossRef CAS PubMed.
- J. X. Jiang, F. Su, A. Trewin, C. D. Wood, H. Niu, J. T. Jones, Y. Z. Khimyak and A. I. Cooper, J. Am. Chem. Soc., 2008, 130, 7710–7720 CrossRef CAS PubMed.
- J. X. Jiang, F. Su, A. Trewin, C. D. Wood, N. L. Campbell, H. Niu, C. Dickinson, A. Y. Ganin, M. J. Rosseinsky, Y. Z. Khimyak and A. I. Cooper, Angew. Chem., Int. Ed., 2007, 46, 8574–8578 CrossRef CAS PubMed.
- T. Ben, H. Ren, S. Ma, D. Cao, J. Lan, X. Jing, W. Wang, J. Xu, F. Deng, J. M. Simmons, S. Qiu and G. Zhu, Angew. Chem., Int. Ed., 2009, 48, 9457–9460 CrossRef CAS PubMed.
- A. P. Cote, A. I. Benin, N. W. Ockwig, M. O'Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166–1170 CrossRef CAS PubMed.
- H. M. El-Kaderi, J. R. Hunt, J. L. Mendoza-Cortés, A. P. Côté, R. E. Taylor, M. O'Keeffe and O. M. Yaghi, Science, 2007, 316, 268–272 CrossRef CAS PubMed.
- F. J. Uribe-Romo, J. R. Hunt, H. Furukawa, C. Klock, M. O'Keeffe and O. M. Yaghi, J. Am. Chem. Soc., 2009, 131, 4570–4571 CrossRef CAS PubMed.
- B. Li, R. Gong, W. Wang, X. Huang, W. Zhang, H. Li, C. Hu and B. Tan, Macromolecules, 2011, 44, 2410–2414 CrossRef CAS.
- M. P. Tsyurupa and V. A. Davankov, React. Funct. Polym., 2002, 53, 193–203 CrossRef CAS.
- M. P. Tsyurupa and V. A. Davankov, React. Funct. Polym., 2006, 66, 768–779 CrossRef CAS.
- R. T. Woodward, L. A. Stevens, R. Dawson, M. Vijayaraghavan, T. Hasell, I. P. Silverwood, A. V. Ewing, T. Ratvijitvech, J. D. Exley, S. Y. Chong, F. Blanc, D. J. Adams, S. G. Kazarian, C. E. Snape, T. C. Drage and A. I. Cooper, J. Am. Chem. Soc., 2014, 136, 9028–9035 CrossRef CAS PubMed.
- D. Wu, A. Nese, J. Pietrasik, Y. Liang, H. He, M. Kruk, L. Huang, T. Kowalewski and K. Matyjaszewski, ACS Nano, 2012, 6, 6208–6214 CrossRef CAS PubMed.
- W. Zhang, F. Liang, C. Li, L. G. Qiu, Y. P. Yuan, F. M. Peng, X. Jiang, A. J. Xie, Y. H. Shen and J. F. Zhu, J. Hazard. Mater., 2011, 186, 984–990 CrossRef CAS PubMed.
- S. Xu, Z. Weng, J. Tan, J. Guo and C. Wang, Polym. Chem., 2015, 6, 2892–2899 RSC.
- J. Yoo, N. Park, J. H. Park, J. H. Park, S. Kang, S. M. Lee, H. J. Kim, H. Jo, J.-G. Park and S. U. Son, ACS Catal., 2015, 5, 350–355 CrossRef CAS.
- Y. Xu, T. Wang, Z. He, A. Zhong and K. Huang, RSC Adv., 2016, 6, 39933–39939 RSC.
- Y. Xu, T. Wang, Z. He, A. Zhong and K. Huang, Microporous Mesoporous Mater., 2016, 229, 1–7 CrossRef CAS.
- M. Arslan, T. N. Gevrek, J. Lyskawa, S. Szunerits, R. Boukherroub, R. Sanyal, P. Woisel and A. Sanyal, Macromolecules, 2014, 47, 5124–5134 CrossRef CAS.
- C. d. Zobrist, J. Sobocinski, J. l. Lyskawa, D. Fournier, V. r. Miri, M. Traisnel, M. Jimenez and P. Woisel, Macromolecules, 2011, 44, 5883–5892 CrossRef CAS.
- M. Benaglia, E. Rizzardo, A. Alberti and M. Guerra, Macromolecules, 2005, 38, 3129–3140 CrossRef CAS.
- J. T. Lai, D. Filla and R. Shea, Macromolecules, 2002, 35, 6754–6756 CrossRef CAS.
- J. Liu, Z. Sun, Y. Deng, Y. Zou, C. Li, X. Guo, L. Xiong, Y. Gao, F. Li and D. Zhao, Angew. Chem., 2009, 121, 5989–5993 CrossRef.
- K. Huang and J. Rzayev, J. Am. Chem. Soc., 2011, 133, 16726–16729 CrossRef CAS PubMed.
- K. Huang, A. Jacobs and J. Rzayev, Biomacromolecules, 2011, 12, 2327–2334 CrossRef CAS PubMed.
- K. Huang and J. Rzayev, J. Am. Chem. Soc., 2009, 131, 6880–6885 CrossRef CAS PubMed.
- F. Rouquerol, J. Rouquerol, K. Sing, P. Llewellyn and G. Maurin, Adsorption by powders and porous solids. Principles, methodology and applications, Academic Press, New York, 2014 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra18836b |
| This journal is © The Royal Society of Chemistry 2016 |