
Facile synthesis of porous CuO polyhedron from Cu-based metal organic framework (MOF-199) for electrocatalytic water oxidation†
 b
and
b
and
Abstract
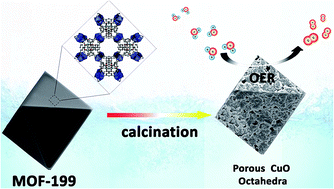
Metal–organic frameworks (MOFs) have been demonstrated as suitable metal sources and sacrificial templates for the preparation of porous transition-metal oxide nanomaterials. Herein, porous CuO micro-polyhedra assembled from numerous nanoparticles are synthesized simply by employing a Cu-based MOF as a precursor via a one-step calcination process under an air atmosphere. Benefitting from their high porosity and large specific surface area, porous CuO materials exhibit excellent electrocatalytic performances toward water oxidation in pH 9.2 KBi solution. A low catalytic onset potential at 1.05 V vs. NHE (NHE = normal hydrogen electrode) is obtained based on electrochemical measurements. A faradaic efficiency of nearly 98% and a stable catalytic current density of 2.2 mA cm−2 are achieved under an applied potential of 1.30 V vs. NHE over an electrolysis period of 10 h. In addition, based on the Tafel plot, the required overpotentials are only ∼410 mV and ∼510 mV for achieving the catalytic current densities of 0.1 mA cm−2 and 1.0 mA cm−2, respectively. The excellent performances make it an appealing candidate as a potential catalyst for green energy applications.
 Please wait while we load your content...
Please wait while we load your content...

