
Selective electrochemical detection of hydroquinone and catechol at a one-step synthesised pine needle-like nano-CePO4 modified carbon paste electrode
Abstract
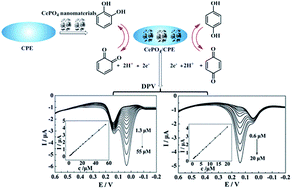
Pine needle-like CePO4 nanomaterials were synthesized by a method of precipitation. X-ray diffraction (XRD), scanning electron microscopy (SEM) and energy dispersive X-ray spectroscopy (EDS) were performed to characterize the nanomaterials. A sensitive and selective electrochemical sensor based on the as-synthesized nanomaterial modified carbon paste electrodes (CePO4/CPE) was successfully constructed and used to study the catalytic oxidation of catechol (CC) and hydroquinone (HQ). The experimental results indicated that the electrochemical responses were improved significantly. Linear calibration ranges were obtained as 0.20–40 μM for CC and 0.40–50 μM for HQ and the detection limits were 0.10 μM and 0.27 μM for CC and HQ, respectively. Moreover, differential pulse voltammetry (DPV) showed that the isomers could be detected sensitively and selectively at CePO4/CPE with peak-to-peak separation about 100 mV, which made possible the selective determination of HQ and CC. The lower detection limit of CC was 0.29 μM and 0.70 μM for HQ was achieved in their binary mixture. Meanwhile, the CePO4/CPE was successfully used for the determination of CC and HQ in local tap water samples and the recoveries were satisfactory. In addition, the proposed method displayed high reproducibility, repeatability, stability and anti-interference performance. Thus, this sensor has potential application in the fields of biomedical and environmental science.
 Please wait while we load your content...
Please wait while we load your content...

