
Mesoporous iron oxide nanowires: synthesis, magnetic and photocatalytic properties†
Abstract
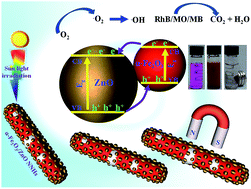
A surfactant- and template-free approach is described for the synthesis of mesoporous α-Fe2O3, Fe3O4 and α-Fe nanowires (NWs). In this approach, α-FeOOH NWs (length 550 nm and diameter 30 nm) were first prepared by hydrolysis of FeCl3. On subsequent thermal treatment in a fluidized bed reactor in the presence of a forming gas (Ar 93% + H2 7%), α-FeOOH transformed to mesoporous NWs of ɑ-Fe2O3, Fe3O4 and ɑ-Fe by controlling the process parameters such as reaction time and temperature. The obtained NWs of α-Fe2O3, Fe3O4 and α-Fe were ferromagnetic at room temperature with a coercive field (Hc) of 412, 583 and 628 Oe respectively. The aligned NWs showed 1.6 to 2 times-enhanced remanence in the parallel direction relative to the perpendicular direction due to magnetic anisotropy. These mesoporous magnetic NWs with a high specific surface area (82 m2 g−1 for α-Fe2O3 NWs) were used in photocatalysis due to the high adsorptivity of three probe dye molecules. The as-prepared α-Fe2O3 NWs exhibited only modest photocatalytic activity; however, the catalytic activity could be further enhanced by decorating the mesoporous ɑ-Fe2O3 NWs with 10 nm sized ZnO nanoparticles. The developed ɑ-Fe2O3/ZnO nanowire nanohybrids could eliminate 100% of the probe dyes: methylene blue, Rhodamine B and methyl orange within 90 min irradiation with solar light, underlining the high photocatalytic degradation efficiency of the nanohybrid. The nanowire nanohybrids could be easily recovered by applying an external magnetic field and reused for at least 4 times without significant loss of their photocatalytic activity.
 Please wait while we load your content...
Please wait while we load your content...

