
Pattern recognition for cytotoxicity mode of action (MOA) of chemicals by using a high-throughput real-time cell analyzer
 *a
*a
Abstract
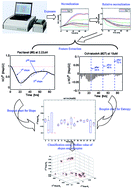
Although the end-point cell-based in vitro assay can provide clear indications of chemical activities, typically fewer clues are given to screen chemicals based on their mode of action (MOA) except reflecting toxicity in a single time-point after living cells are exposed to those chemicals. To determine distinct chemical effects characterized by MOA, a statistical pattern recognition analysis using multi-concentration time-dependent cellular response profiles (TCRPs) has been developed. By monitoring the dynamic cytotoxicity response profile of living cells via the xCELLigence RTCA HT system, changes in cell number (termed as CI) caused by different MOAs are recorded on-line as a time series. By comparing the cellular response of the treated cells to untreated cells (terms as negative control), a relative normalized cell index (NCIR) is presented which achieves the same baseline for in vitro assay and reduces variation caused by different experiments. Dynamic features, which reflect the cell killing, cell lysis, cellular proliferation and certain cellular pathological changes, are extracted from TCRPs. Finally, a hierarchical clustering analysis integrated with k-means clustering algorithm is employed to classify the extracted features, and develop MOA assays to detect and distinguish chemicals. The proposed method, including feature extraction, statistical analysis and clustering algorithm, can be readily automated and enables relatively high throughput screening for MOA hits at the cellular level.
 Please wait while we load your content...
Please wait while we load your content...

