
Arene ruthenium(ii) complexes with chalcone, aminoantipyrine and aminopyrimidine based ligands: synthesis, structure and preliminary evaluation of anti-leukemia activity†
Abstract
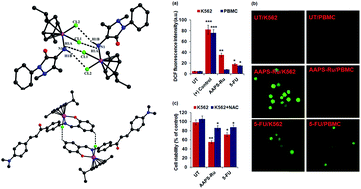
A series of arene ruthenium(II) (Ru) complexes with N-monodentate (AAP) and N,O- and N,N-bidentate chelating ligands (AAPS, ADABS, AAPPA and P2P) have been synthesized and evaluated for preliminary antileukemia activity. Single crystal X-ray studies of AAP-Ru, ADABS-Ru and P2P-Ru indicates that Ru(II) adopted a typical “three-leg piano-stool” geometry with pseudo-tetrahedral (AAP-Ru) and quasi-octahedral arrangements (ADABS-Ru and P2P-Ru). Supramolecular interactions (C–H⋯Cl, N–H⋯Cl and π–π) in the crystal lattice of AAP-Ru and ADABS-Ru resulted in a dimeric structure whereas P2P-Ru showed a supramolecular 1D chain. Importantly, preliminary in vitro anticancer activities for Ru-arene complexes have been evaluated against K562 (human chronic myeloid leukemia cell line) by means of MTT assay that showed strong anti-leukemic activity for AAPS-Ru (IC50 = 12.46 ± 0.47 μM) and P2P-Ru (IC50 = 11.8 ± 0.49 μM). The mechanistic studies indicated reactive oxygen species (ROS) mediated selective leukemic cells death without affecting the normal cells by AAPS-Ru. The initiation of apoptosis by AAPS-Ru treatment, particularly TNF-α induced cell death, was confirmed by pre-treatment with a selective TNF-α inhibitor.
 Please wait while we load your content...
Please wait while we load your content...

