
An artful and simple synthetic strategy for fabricating low carbon residual porous g-C3N4 with enhanced visible-light photocatalytic properties†
Xiaopeng Hana,
Yuexin Wanga,
Jianan Lva,
Lingru Konga,
Li Tiana,
Xuemei Lua,
Jiwei Wang*ab and
Xiaoxing Fan*ab
aSchool of Physics, Liaoning University, Shenyang, 110036, P. R. China. E-mail: xxfan@lnu.edu.cn
bLiaoning Key Laboratory of Semiconductor Light Emitting and Photocatalytic Materials, Liaoning University, Shenyang 110036, P. R. China. E-mail: jiweiwang6688@yahoo.com
First published on 29th August 2016
Abstract
Nanoporous graphitic carbon nitrides (g-C3N4) show great photocatalytic performance owing to their unique structure. Organic templating usually was used to increase the porosity of g-C3N4, but the templating method is restricted, the technical problem is that the condensation towards larger extended CN-structures takes place around or above the decomposition temperature of the template, and thus, causes the resealing of the pores. Furthermore, the templating method usually leads to high carbon residue and lowers the photocatalytic performance. Here, the issue is addressed through the development of an artful and simple synthetic strategy to avoid the decomposition of the template at too early a stage, by taking advantage of the “two-step” heat-treated process which firstly heated in an N2 atmosphere then calcined in air. Also, the two-step strategy is easily used for obtaining low carbon residual g-C3N4 with enhanced photocatalytic activity. The obtained samples possess a higher specific surface area (126 m2 g−1) and more abundant pores, and a band gap increase of 0.08 eV in comparison with bulk g-C3N4. The photocatalytic activity of the samples was evaluated by oxidative dehydrogenation of 2-propanol to acetone under visible light illumination. The enhanced performance was mainly ascribed to the high specific surface area, which contains more active sites for reaction. This artful and simple strategy can in general be expanded to the organic templating method for preparing porous g-C3N4, enabling the creation of stable polymeric CN nanostructures with maximized material and structure functions.
Introduction
Photocatalysis holds great promise for addressing global energy and environmental problems. Extensive studies have been devoted to exploiting highly-efficient semiconductor photocatalysts that are photoactive in H2 evolution and organic pollutant degradation under visible light irradiation.1,2 Recently, polymeric graphitic carbon nitride (g-C3N4), as a metal-free photocatalyst, has attracted enormous attention owing to the superior photoelectrochemical properties with thermal and chemical stabilities under ambient atmosphere.3 Also g-C3N4 has a suitable energy band gap (2.7 eV) with conduction band (−1.3 eV) and valence band (1.4 eV) potentials vs. NHE.4 Moreover, g-C3N4 exhibits an appropriate microstructure with unsaturated nitrogen atoms for anchoring active sites.5 Due to these superior properties, g-C3N4 has been proven to have promising applications in sustainable energy fields as a heterogeneous metal-free catalyst for solar water splitting, carbon dioxide reduction and environmental cleaning under visible light irradiation.6,7 Furthermore, g-C3N4 is earth-abundant and easily synthesized by thermal condensation of cheap and nitrogen-rich precursors (such as urea, thiourea, melamine, dicyandiamide, cyanamide).3,6,8–10 However, the quantum efficiency of g-C3N4 is still low, which is mainly attributed to its weak visible-light absorption, high charge recombination, and small surface area.3,11 So many approaches, such as doping with foreign elements,12–15 surface modification,16,17 heterostructured nanocomposites fabrication,18–20 and so forth, have been employed to improve the photocatalytic performance of g-C3N4.The synthesis of porous g-C3N4 with high specific surface area, small particle size, and tunable pore diameter is attracting more attention because a larger specific surface area of photocatalysts can be favorable for photocatalytic reaction by providing abundant reactive sites and short bulk diffusion length to reduce the recombination probability of photoexcited charge carriers.21,22 The synthesis methods of porous g-C3N4 can be broadly classed as “hard” template and “soft” template methods. The hard template typically employs solid porous materials, such as porous anodic aluminium oxides or porous silica, for nanocasting of g-C3N4 in which a precursor such as dicyandiamide was filled and heated to produce g-C3N4.23,24 The hard templating method is very effective, giving specific surface area up to 517 m2 g−1.25 However, the templates have to be removed in an extra step involving aqueous ammonium bifluoride (NH4HF2) or hydrogen fluoride (HF), which are hazardous and not environment-friendly and difficult to further functionalize.26
Compared with the hard template approach, the “greener” soft template route not only simplifies the entire synthetic procedure but also allows for easy tuning of the morphology through choice of different templates.27 Some surfactants and block polymers as soft templates used for the production of nanoporous materials. However, due to thermodynamic and physicochemical reasons, it is sometimes still a challenge for using these soft templates to prepare porous g-C3N4. The practical problem is that the condensation towards larger extended CN-structures takes place around or above the decomposition temperature of the common used template.28 Wang and co-workers succeed in obtaining larger specific surface area g-C3N4 with various soft templates such as Triton X-100, P123, F127, Brij30, Brij58 and Brij76.29 Their success can be attributed to the high temperature tolerated surfactant (Triton X-100) or complicated thermal treatment avoiding a closed pores system. However, the high carbon residue in these materials interrupts the structure of g-C3N4 and lowers the photocatalytic performance.27 Clearly, to address these issues, it is desirable to explore a new synthetic strategy that can effectively avoid a closed pore system, as well as keep the low carbon residue has become a rather urgent necessity to obtain helpful porous g-C3N4 photocatalysts.
Here, we proposed a strategy can obtain low carbon residual and large surface area g-C3N4, by employing acetic acid mediated melamine. The acetic acid mediated melamine has been used for synthesizing g-C3N4 before, but the reported specific surface area is only 12.3 m2 g−1, and no mechanism was highlighted.30 Based on our results, it is believe that the low specific surface area is due to the pore resealing during the calcination caused by the carbon residue removal at too early stage before the CN structure formation. For resolving this problem, an artful and simple synthetic strategy focused on delay pore-forming project was designed according a two-step thermal treatment process. That is to say, first step is heating the acetic acid treated melamine at N2 atmosphere, thus keeping carbon residue from the composition of templating exist in the g-C3N4, then further calcination in air to remove the carbon in air and obtain large specific surface area g-C3N4. The as-prepared porous g-C3N4 shows much higher BET specific surface area and highly enhanced photocatalytic performance than pure g-C3N4. Moreover, a series of experiments are designed to clarify the mechanism of porous g-C3N4. More important is this strategy can be expand to other system which organic acid treatment melamine for synthesizing porous g-C3N4.
Experimental
Synthesis of porous and bulk g-C3N4
The synthesis of porous of g-C3N4 is as follow: melamine (2.52 g) was dissolved in distilled water (100 ml) then heated at 80 °C for 0.5 h. Subsequently, a certain amount of acetic acid (1.15 ml, 2.30 ml, 3.45 ml and 4.60 ml, the molar ratios of acetic acid to melamine are 1, 2, 3 and 4, respectively) was added to the above melamine solution and stirred at 80 °C for 2 h. The white powder was collected by drying the above solution at 100 °C for 12 h. The acetic acid treated melamine was placed in an alumina crucible with a cover and then put into the middle region of a quartz tube. The quartz tube was heated to 550 °C in a tubular furnace for 4 h at a rising rate of 5 °C min−1 with N2 flow of 40 ml min−1. The calcined products are abbreviated to g-CN-1, g-CN-2, g-CN-3 and g-CN-4, according to the molar ratios of acetic acid to melamine, respectively. For example, g-CN-3 means g-C3N4 starting with 3 of acetic acid/melamine mol ratio. Then, the samples of g-CN-1, g-CN-2, g-CN-3 and g-CN-4 were further calcined at 550 °C for 2 h in air, and were abbreviated to g-CNA-1, g-CNA-2, g-CNA-3 and g-CNA-4, respectively (A denotes heating in air atomosphere).Bulk g-C3N4 was prepared at the same process above-mentioned but without acetic acid treatment. The pure bulk g-C3N4 was abbreviated to g-CN-0, and g-CNA-0 (further calcined in air), respectively.
Characterization
The thermogravimetric-differential scanning calorimetry analysis (TG-DSC) measurements were performed on NETZSCH STA 409 PG/PC analyzer, the detected range of temperature is from room temperature to 800 °C at a heating rate of 10 °C min−1. X-ray diffraction (XRD) patterns were obtained using a powder X-ray diffractometer (Cu Kα radiation source, DX-2700, Tongda Co. Ltd. China). The UV-vis diffuse reflectance spectrum was recorded by a UV-vis spectrophotometer (UV-2550, Shimadzu) at room temperature and transformed to the absorption spectrum according to the Kubelka–Munk relationship. The nitrogen adsorption and desorption isotherms were measured via nitrogen physisorption at 77k on a surface area and porosity analysizer Instrument (Quadrasorb evo, Quantachrome). The Fourier transform infrared (FT-IR) experiment was obtained using a Magna-IR750 FT-IR spectrometer in the KBr pellet, scanning from 4000 to 400 cm−1. The morphology of the powders was examined by a field emission scanning electron microscopy (S4800, Hitachi). TEM images were obtained on a JEOL JEM-2100 electron microscope with an accelerating voltage of 200 kV, the samples for TEM were prepared by dispersing the final powders in ethanol, and the dispersion was dropped on carbon–copper grids. X-ray photoelectron spectroscopy (XPS) data were obtained on a PHI 5000 Versa Probe using 200 W monochromated Al Kα radiation. Binding energies were calibrated using the adventitious carbon (C 1s) = 284.6 eV. The atomic ratio of each element in different samples was determined via elemental analysis (CHN-O-Rapid, Heraeus).Photocatalytic activity measurements
The photocatalytic activities of the samples were performed by oxidizing 2-propanol using a gastight system with a quartz window. In a typical process, powder sample (0.1 g) was put on a 4 cm2 glass groove. The glass with photocatalyst was then put into a 300 cm2 reactor, filled with air to one atmospheric pressure. Then, 5 μl 2-propanol solutions were injected into the reactor to generate a high-concentration 2-propanol gas. The light source for the catalytic reaction was a 300 W Xe arc lamp. Acetone and 2-propanol were detected by a gas chromatography with FID detector (GC1690, JieDao Tech).Results and discussion
In order to understand the phase transformation process of the g-CN-3 and g-CNA-3, TG-DSC was carried out. The DSC and TG thermograms for the acetic acid treated melamine (the precursor of g-CN-3) were shown in Fig. 1a. Below 120 °C, the weight loss was ascribed to volatile species (including water and CH3COOH). Between 120 °C and 330 °C, a mass loss arised from the dehydration and the decomposition of amide group (–NH–CO–CH3, formed by the reaction of –NH2 and CH3–COOH), which would transform to residual carbon species during the heating in nitrogen atmosphere. As a result, an exothermic reaction peak (295 °C) was observed because of the dehydration of amide groups. The strongest endothermal peak with a rapidly weight loss appears in the temperature range 330–390 °C, which indicates that the melamine becomes unstable and no residue of the material is observed. This phenomenon can be attributed to the sublimation and thermal condensation of melamine in an open system. The transformation of g-CN-3 to g-CNA-3 was detected in air atmosphere, as shown in Fig. 1b, a slight weight loss appeared companying with a broader exothermic peak before 550 °C. It may be explained by that residual carbon species in g-CN-3 reacted with oxygen to generate heat. A rapid weight loss around 600 °C took place due to decomposition of g-CN-3, and when heated to 750 °C all weight loss occurred. The DSC curve also indicates that decomposition of the as-prepared g-CNA-3 occurred first at 600 °C since a big exothermic peak appears due that the decomposition products burn immediately.31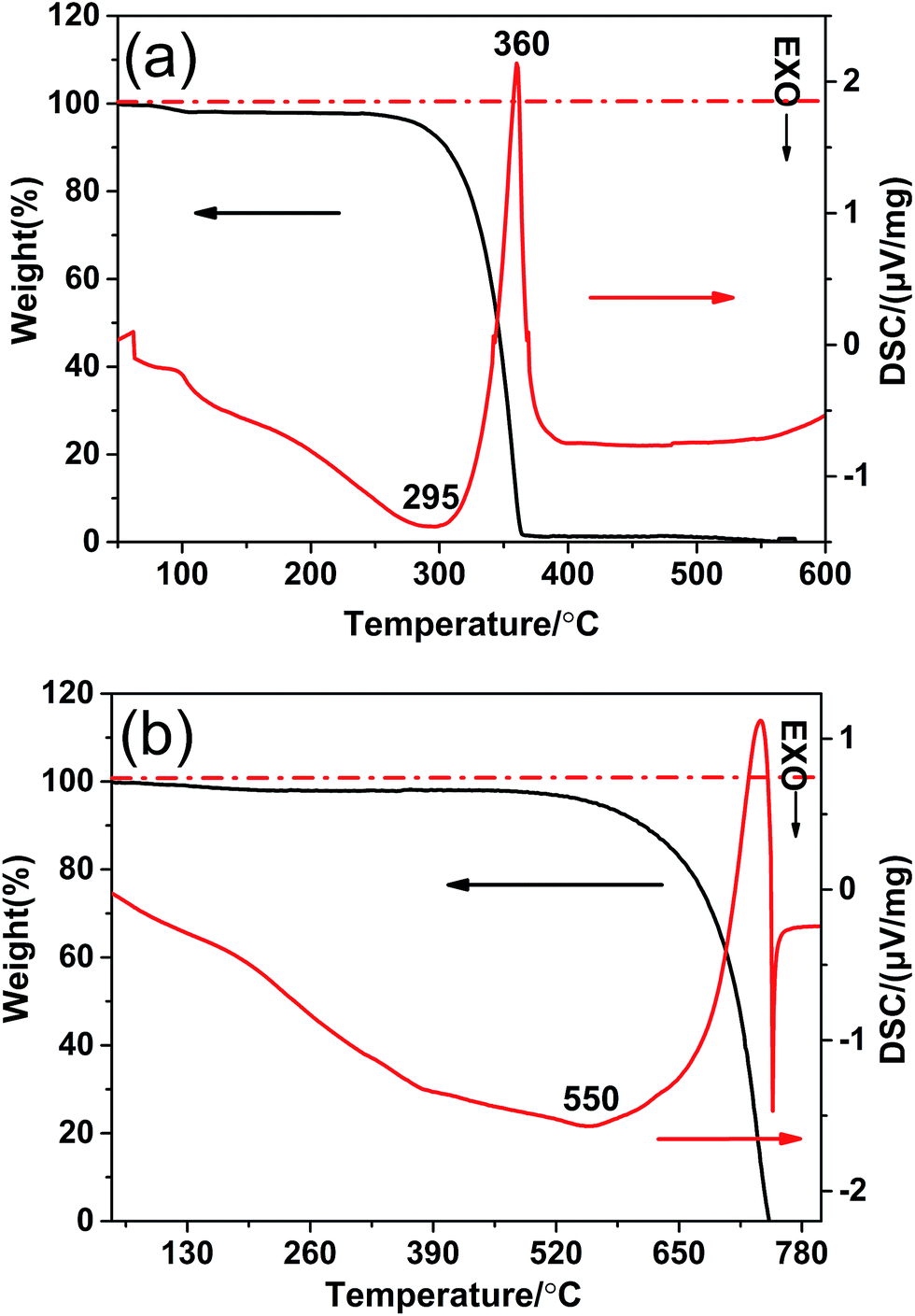 | ||
| Fig. 1 TG-DSC thermograms for heating (a) the acetic acid treated melamine in N2 and (b) the g-CN-3 in air. | ||
XRD was performed to characterize the phase structure of the products. Fig. 2 shows the XRD patterns of g-CN-0, g-CNA-0, g-CN-3 and g-CNA-3. All XRD patterns exhibit two distinct peaks at 27° and 13°, which respectively correspond to the characteristic diffraction of the (100) and (002) planes of the g-C3N4, and caused by the interlayer stacking of aromatic segments and the in-plane structural packing motif, respectively. The similarity of XRD patterns indicates that acetic acid has no significant influence on the formation of graphitic stacking structures of g-C3N4, and the most of NH2 group of melamine have been reserved. Compared with g-CN-0, the (002) peak of g-CNA-0 lightly shifts towards a high angle, this implies the decreasing interlayer distance in the g-C3N4,32 which can be explained by that the undulated single layer in g-C3N4 was planarized by heating in air, leading to a denser stacking and decreasing the interlayer distance.21,33 The average crystallite sizes were calculated to be 8.9, 9.5, 6.4 and 6.7 nm for g-CN-0, g-CNA-0, g-CN-3 and g-CNA-3 according to the broadening of the (002) peak in the XRD patterns by Scherer's formula, respectively. Compared with g-CN-0, the crystal size of g-CNA-0 increased after further heating in air, this can be ascribed to the high temperature which enhanced the grains growth. The comparison of g-CNA-0 and g-CNA-3 shows that the utilization of acetic acid leads to the decrease of crystallite size and the inhibition of the crystal growth of graphitic carbon nitride. This phenomenon also can be found in the XRD patterns of g-CN-1, g-CNA-1, g-CN-2, g-CNA-2, g-CN-4, g-CNA-4 (Fig. S1†).
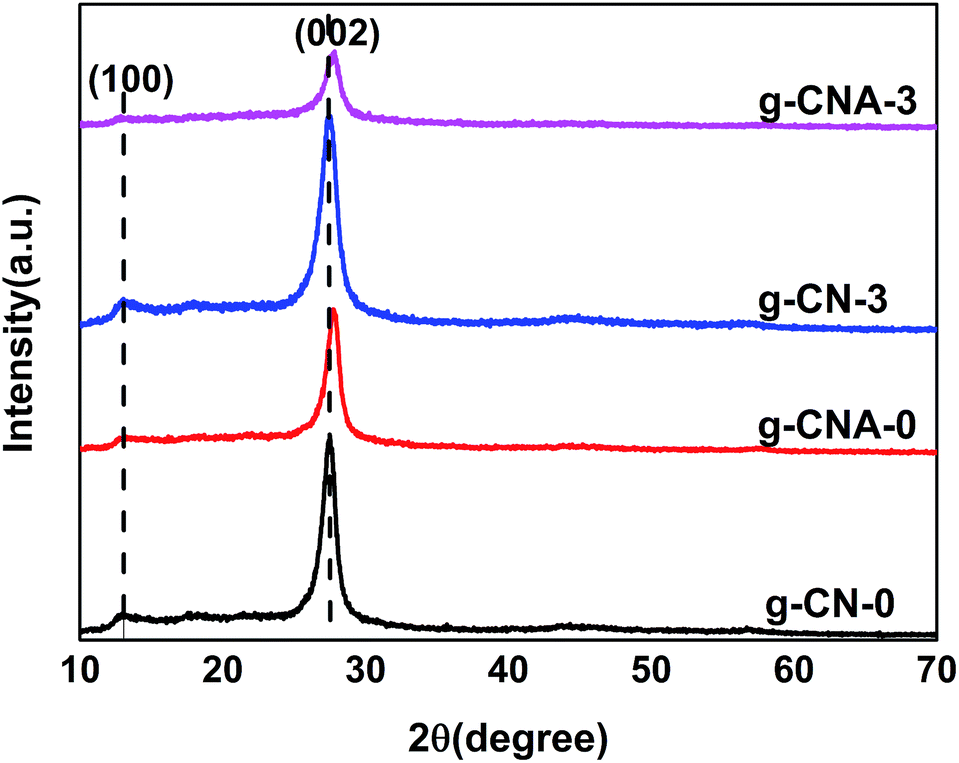 | ||
| Fig. 2 XRD patterns of g-CN-0, g-CNA-0, g-CN-3 and g-CNA-3. | ||
The porous architecture is confirmed by N2 adsorption–desorption isotherms and corresponding pore size distribution (Fig. 3). The isotherms exhibit type III behavior, which is caused by the weak adsorbent interaction. And all samples show type H3 hysteresis loops at 0.45 < P/P0 < 0.998 in the isotherms, which imply the existence of porous structures in the sample and are often observed on the aggregates of plate-like particles giving rise to slit-shaped pores.34 It should be noted that the specific surface area of g-CN-0 synthesized without using acetic acid is 9 m2 g−1, and it increases to 51 m2 g−1 after 2 h calcination at 550 °C in air, the increasing in surface area of the g-C3N4 is mainly caused by the thermal oxidation etching in air. The specific surface areas are 99, 112, 126 and 74 m2 g−1 for g-CNA-1, g-CNA-2, g-CNA-3 and g-CNA-4, respectively. All samples synthesized by acetic acid treated melamine show larger specific surface area than those from pure melamine. Increasing the number of acetic acid leads to an increase in the specific surface area, this is mainly due to the increased number of pore-makers by which help form more channels. When the ratio of acetic acid to melamine was high, the surface area was low on g-CNA-4 that could be a result of the pore collapse during heating process on account of the presence of excess amounts of carbon species. The g-CNA-3 possesses a supreme specific surface area (126 m2 g−1), while the specific surface area of g-CN-3 is 12 m2 g−1 which is much lower than that of g-CNA-3 (Fig. S2 and Table S1†). Based on above data, it is believed that the high specific surface area of g-CNA-3 may be mainly contributed by three factors. Firstly, the amide groups and formed carbon species inhibit the subsequent thermal condensation and the crystal growth, thus improve the specific surface area. Secondly, some pores formed by removing carbon species can contribute to the specific surface area greatly. Furthermore, the formed pores help the oxygen entering the inner of the g-C3N4, enhancing the thermal oxidation etching and increasing the specific surface area further.21 It is observed that a wide pore size distribution ranging from 2 to 28 nm for g-CNA-X (X = 1, 2, 3 and 4), demonstrating that acetic acid mediation could be used to construct mesoporous structures with significant increased specific surface area to promote photocatalytic activity.
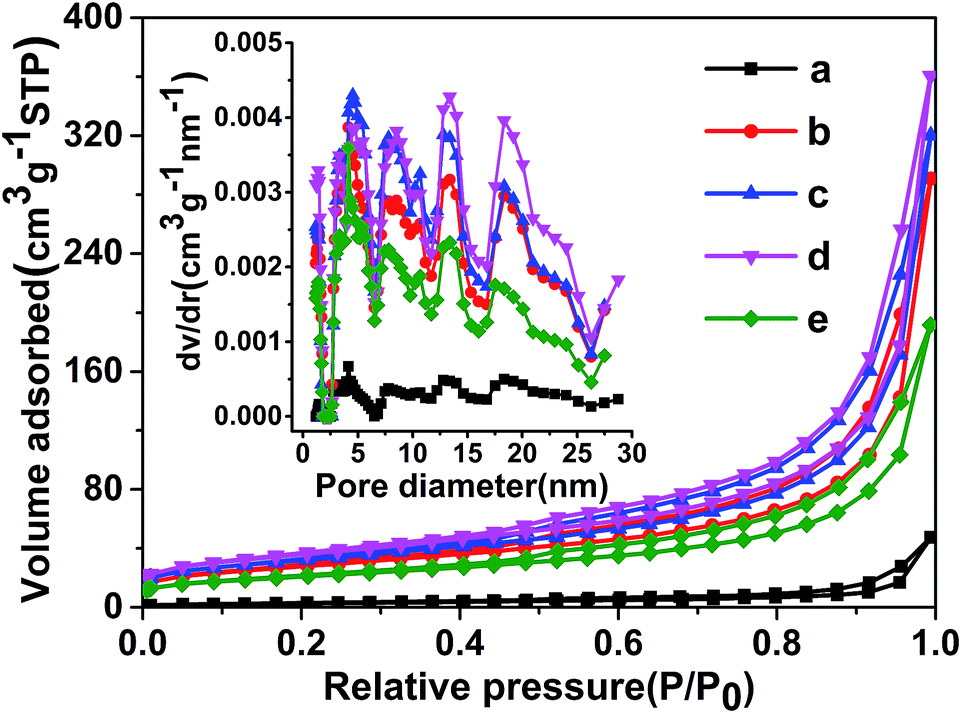 | ||
| Fig. 3 Nitrogen adsorption/desorption isotherm and corresponding pore size distribution of porous g-C3N4 prepared with different of acetic acid: (a) g-CN-0, (b) g-CNA-1, (c) g-CNA-2, (d) g-CNA-3, (e) g-CNA-4. | ||
The morphology and microstructure of samples were investigated with SEM as shown in Fig. 4. Compared to the g-CN-0 consisting of stratiform solid agglomerates with smooth and flat, the g-CN-3 presents smaller particle, this indicates that the treatment of acetic acid inhibits the conjunction of melamine and leads to decreasing the grain size of g-C3N4, in accordance with the XRD patterns. The g-CNA-0 shows some small agglomerates with coarse surface, while g-CNA-3 becomes a loose, thin and faveolate feature with some pores on the layer surface when oxidized by air. Moreover, TEM image of pure g-C3N4 illustrates a typical low porosity and platelet-like structure (Fig. 4e) but that of g-CNA-3 shows the small flat sheet mixed with white void spaces (Fig. 4f). The white void presented irregular shape is corresponding the pore. It can be seen plenty of irregular pores existed and the pore diameter is about several tens nanometre, which is consistent with the result of BJH pore size distribution. Apparently, these results confirm the high specific surface area and large pore volume of porous g-C3N4 can be obtained by this method.
 | ||
| Fig. 4 SEM images of (a) g-CN-0, (b) g-CNA-0, (c) g-CN-3 and (d) g-CNA-3; TEM images of (e) g-CN-0 and (f) g-CNA-3. | ||
XPS and elemental analysis were used to investigate the chemical compositions of the final products.
Table 1 summarizes the surface and bulk C/N atomic ratio measured by XPS and elemental analysis, respectively. As known, the ration of C/N should be 0.75 for a perfect g-C3N4 crystal, however, the elemental analysis results show that the C/N ratio of g-C3N4 prepared by directly heating melamine (g-CN-0) is 0.67 which is less than 0.75, this phenomenon also be reported in other articles,22 the reason is because there are un-condensed amino groups in the materials. The stretching mode of NH2 groups in FT-IR spectra also confirms the existence of surplus amino groups (Fig. S3†) in g-CNA-3. Compared to g-CN-0, the increase in C/N atomic ratio of g-CNA-3 indicates using more acetic acid leads to more carbon residue, which also can be found in other samples (Table S2†). The C/N atomic ratio of g-CNA-X (X = 1, 2, 3 and 4) is close to that of g-CN-0, and the ratio of C/N of g-CNA-3 decreases compared with g-CN-3, both indicating that the carbon residue was removed by calcination in air.
| g-CN-0 | g-CNA-0 | g-CN-3 | g-CNA-3 | |
|---|---|---|---|---|
| XPS | 0.87 | 0.77 | 0.89 | 0.75 |
| EA | 0.67 | 0.67 | 0.68 | 0.67 |
From the XPS data, it can be seen besides the main elements of C and N on the surface (Fig. 5a and b), there is a small amount of oxygen, which is mainly attributed to the adsorbed water and carbon dioxide. The surface C/N atomic ratio of g-CN-0 is 0.87, which is higher than that of g-CN-0. The higher surface C/N atomic ratio can be caused by that a certain amount of surface nitrogen was disappear during condensation and that sample absorbs containing carbon pollutant.35 In addition, XPS results also show a likely law that the C/N atomic ratio decreased when calcined in air, for instance, g-CN-3 is 0.89, but decreased to 0.75 for g-CNA-3, this is in accordance with the results of elemental analysis. The high-resolution XPS spectra of C 1s were selected to further investigate the surface chemical compositions of different samples. As shown in Fig. 5c, e and S4a,† the XPS spectra of C 1s for all samples can be fitted into three peaks, the peaks at 284.6, 285.85 and 288.1 eV are attributed to C–C, C–NH2 and N–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N, respectively.36 The C atom content of 284.6 eV is 9.5% and 18% for g-CN-0 and g-CN-3 respectively, indicating that introducing acetic acid leads to the carbon species residue originated of the dehydration of amide groups. It decreased to 2.6% for g-CNA-3 when calcined in air, this is caused by that the carbon species removed by oxidation of air. The N 1s spectrum of all simples can be mainly deconvoluted into four peaks with binding energies at about 398.5 eV, 399.1 eV, 400.9 eV and 404.2 eV respectively (Fig. 5d, f and S4b†). The dominant peak at 398.5 eV corresponds to the sp2-hybridized nitrogen in C containing triazine rings (C–NC), whereas the peak located at 399.1 eV is usually attributed to the bridging N atoms in N–(C)3 groups. The peak at 400.9 eV indicates the amino groups (N–H), and the peak at 404.2 eV is attributed to charging effects. Notably, the peak at 399.1 eV becomes weak, meanwhile, a new peak at 399.9 eV ascribing to H–N–(C)2, appears in the N 1s spectra of g-CN-3. These changes confirm that the triazine rings in g-CN-3 exists part connection through two-coordinated bridging nitrogens. So, it is concluded that the condensation modal of melamine changed since three-coordinated bridging nitrogens partly transformed to two-coordinated bridging nitrogens, which also be found in the condensation of hydrochloric acid treated melamine.37
N, respectively.36 The C atom content of 284.6 eV is 9.5% and 18% for g-CN-0 and g-CN-3 respectively, indicating that introducing acetic acid leads to the carbon species residue originated of the dehydration of amide groups. It decreased to 2.6% for g-CNA-3 when calcined in air, this is caused by that the carbon species removed by oxidation of air. The N 1s spectrum of all simples can be mainly deconvoluted into four peaks with binding energies at about 398.5 eV, 399.1 eV, 400.9 eV and 404.2 eV respectively (Fig. 5d, f and S4b†). The dominant peak at 398.5 eV corresponds to the sp2-hybridized nitrogen in C containing triazine rings (C–NC), whereas the peak located at 399.1 eV is usually attributed to the bridging N atoms in N–(C)3 groups. The peak at 400.9 eV indicates the amino groups (N–H), and the peak at 404.2 eV is attributed to charging effects. Notably, the peak at 399.1 eV becomes weak, meanwhile, a new peak at 399.9 eV ascribing to H–N–(C)2, appears in the N 1s spectra of g-CN-3. These changes confirm that the triazine rings in g-CN-3 exists part connection through two-coordinated bridging nitrogens. So, it is concluded that the condensation modal of melamine changed since three-coordinated bridging nitrogens partly transformed to two-coordinated bridging nitrogens, which also be found in the condensation of hydrochloric acid treated melamine.37
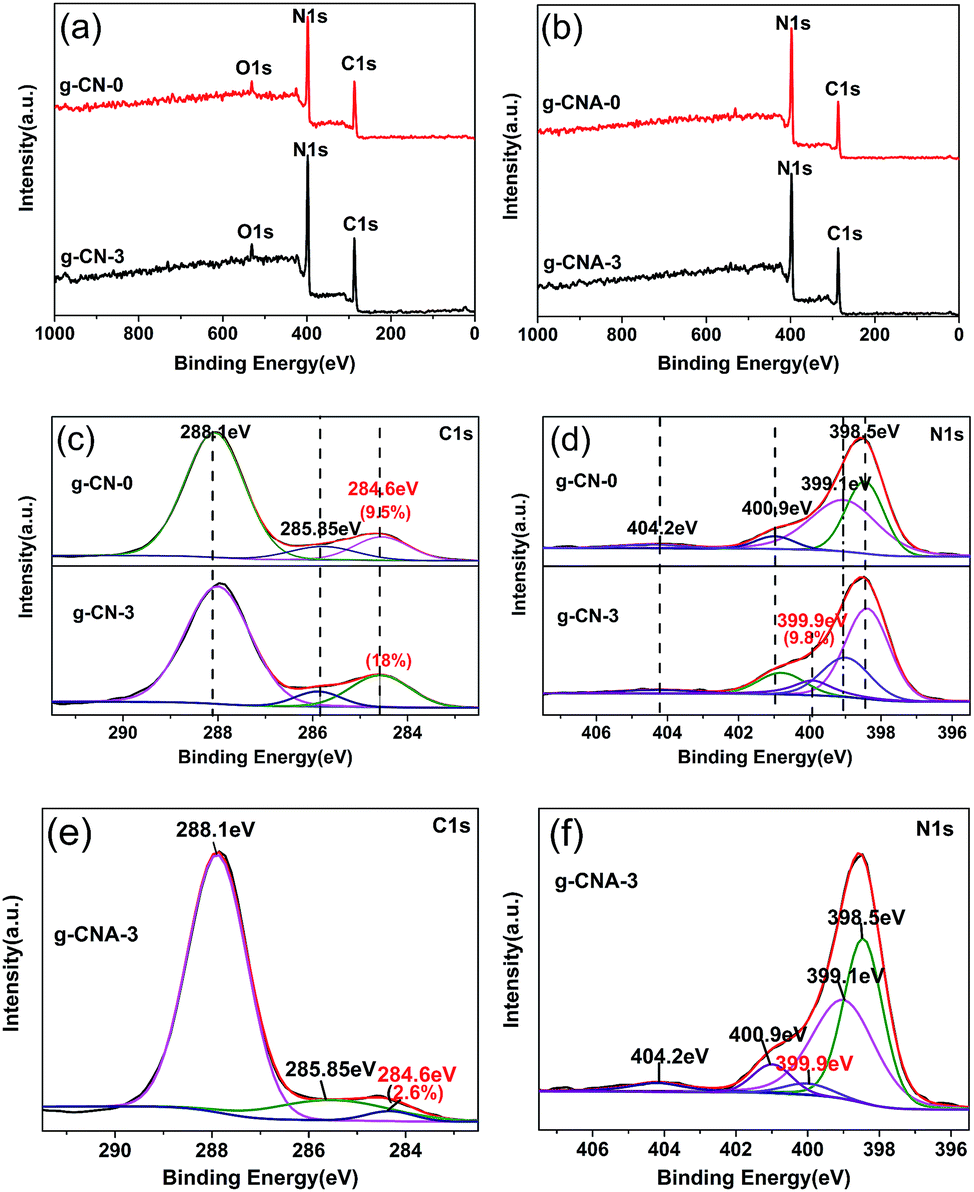 | ||
| Fig. 5 XPS spectra of the resulting samples: (a) survey of g-CN-0 and g-CN-3, (b) survey of g-CNA-0 and g-CNA-3, (c) C 1s spectra of g-CN-0 and g-CN-3, (d) N 1s spectra of g-CN-0 and g-CN-3, (e) C 1s spectra of g-CNA-3 and (f) N 1s spectra of g-CNA-3. | ||
Based on the above results, the possible mechanism of obtaining larger specific surface area g-CNA-3 can be explained as follows. As illustrated in Scheme 1, when heating, acetic acid would react with part of the amino groups of melamine through an intermolecular dehydrolysis reaction, and amide (–NH–CO–) formed by the reaction of –NH2 and –COOH.38 It should be noted that it is an incompletely reaction, not all amino groups reacted with acetic acid, because no obviously change in FT-IR absorbed curves (Fig. S5†) between acetic acid treated melamine and pure melamine can be found. The remained –NH2 participates in the subsequent thermal condensation, while the amide would inhibit the condensation, leading to a certain amount of voids formation near the amide groups sites. Meantime, part of bridging nitrogens in g-C3N4 changed from three-coordinated bridging nitrogens to two-coordinated bridging nitrogens.37 At high temperature in N2 atmosphere, the amide or other uncondensed large molecular decomposed and remained as carbon residues which were detected by the XPS. The carbon residues not only inhibit the growth of grains, but also act as pore-maker. Therefore, the role of acetic acid is help to form the precursor of carbon residue. Finally, porous g-C3N4 was obtained by further heating to remove the residual carbon species and allowing the thermal oxidation etching occurring inside. It is believed that the obtained nanoporous structure is contributed by two parts. One is from the removing of carbon residue because more microporous pore can been found in pore size distribution. The rest is come from the thermal oxidation etching which would enlarge the pores. The micropores formed by carbon residue is very important to help the oxygen entering the inner of the g-C3N4 and allowing the thermal oxidation etching occurring inside. The size of pores formed by thermal oxidation etching should be depended on the time of etching, and is as large as several tens nanometer under our synthesis condition. The success of obtaining larger surface g-C3N4 is due to adopting a two-step heating treatment. The first thermal treatment at N2 atmosphere makes the melamine condensation and leaves residual carbon in g-C3N4. The second one aims at removing the residual carbon, releasing the pores and occurring thermal oxidation etching. If adopting one-step heating, neither in N2 atmosphere nor in air can succeed. It can be found only N2 thermal treatment samples, for example g-CN-3, usually are characterized by high carbon content and small specific surface area (12 m2 g−1), while simply calcination at air would cause a closed pore system with small specific surface area (12.3 m2 g−1, reported by Wu and co-workers),30 as decomposition of pore-maker at too early stage enables the pore resealing of the pores.
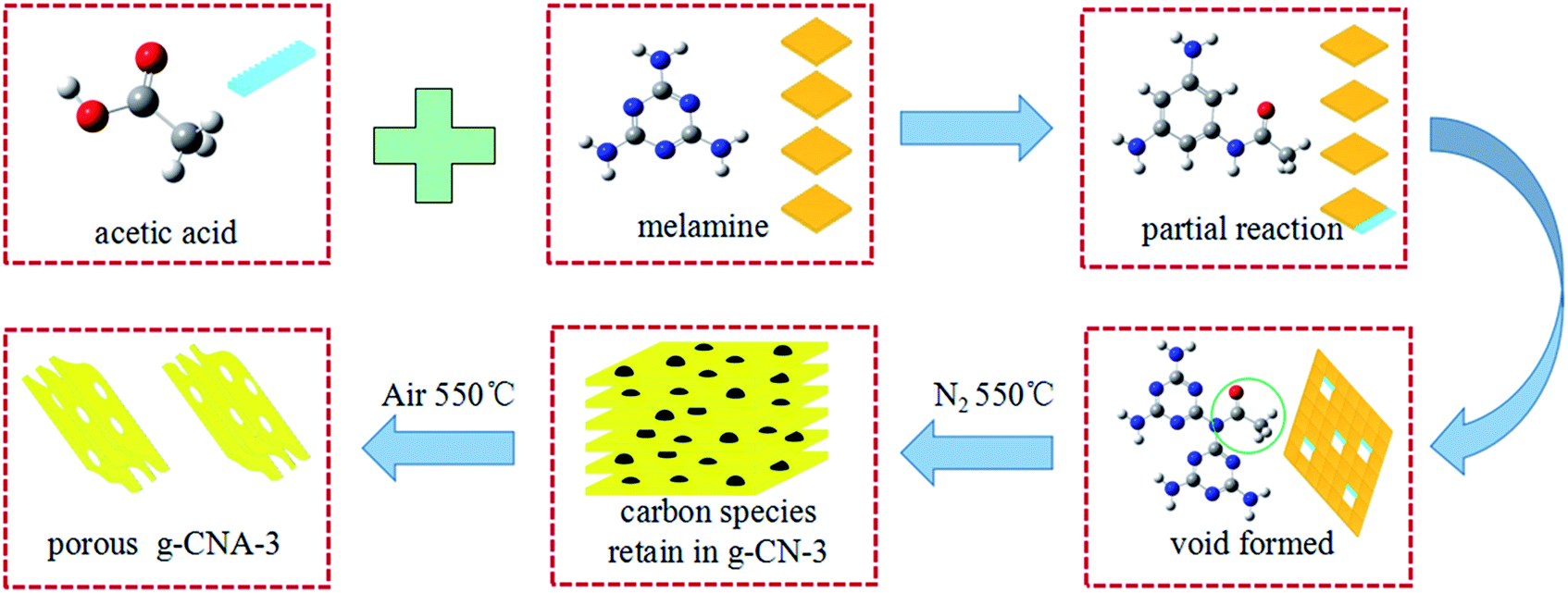 | ||
| Scheme 1 Schematic illustration of the synthesis process of porous g-CNA-3. | ||
The UV-vis diffuse reflectance spectra of the final samples are presented in Fig. 6a. All the UV-vis absorption edges of g-CNA-0, g-CNA-1, g-CNA-2, g-CNA-3 and g-CAN-4 showed a blue shift compared with that of g-CN-0. This occurrence could be attributed to the quantum size effect because of smaller crystal sizes and porosity. The band gap energy (Eg) can be estimated from the intercept of the tangents to the plots of (αhν)1/2 versus photon energy, as shown in Fig. 6b. The band gaps optically obtained were approximately 2.78, 2.90 and 2.86 eV for the samples of g-CN-0, g-CAN-0, and g-CAN-3, respectively. The larger band gap by shifting conduction and valence bands in opposite directions can contribute to a stronger redox ability of charge carriers and prolonging the lifetime of charge carriers.21 Through the quantum size effect limits the visible light absorption, which is a necessary result of increasing specific surface area of sample, because the wall of large specific surface area structure usually construct by small nanocrystals, it is significant to develop large specific surface area photocatalysts, as increasing the surface area can greatly enhance the photocatalytic activity even absorbing less light. In addition, samples from acetic acid mediated melamine show a slight absorption at the range from 450 nm to 600 nm, which could be related to the increased defects.32
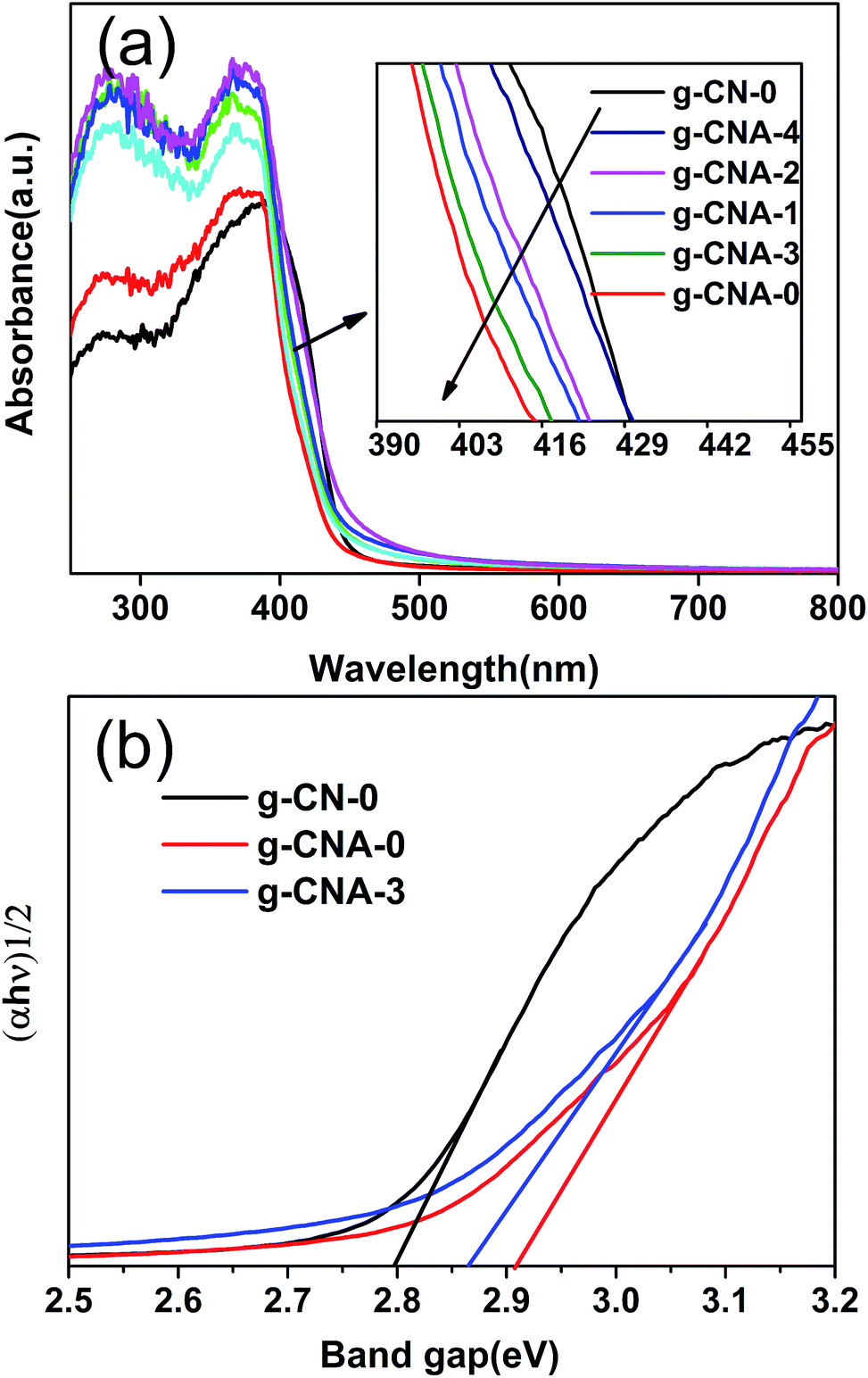 | ||
| Fig. 6 UV-vis absorption spectra of (a) g-CN-0, g-CNA-0, g-CNA-1, g-CNA-2, g-CNA-3 and g-CNA-4; (b) the transformed Kubelka–Munk function vs. the energy of the light absorbed of g-CN-0, g-CNA-0 and g-CNA-3. | ||
The photocatalytic activities of the samples were evaluated by oxidative dehydrogenation of 2-propanol to acetone under visible light irradiation (λ > 420 nm). 2-Propanol photodegradation is firstly by photo-oxidative dehydrogenation to give acetone and eventually photo-oxidization to CO2. Since the self-oxidation of 2-propanol is negligible, 2-propanol photodegradation can serve as a good reaction model to evaluate the photocatalytic activities of semiconductors.39 Fig. 7a shows the concentration of evolved acetone over different photocatalysts under visible light irradiation. Acetone can be evolved over all the tested catalysts under light irradiation, but it is obvious that the photocatalytic activities for all of the g-CNA-X samples obtained from acetic acid mediate melamine are higher than that of g-CN-0 obtained from melamine. In order to investigate the reaction rate, evolved rate of acetone over different catalysts were also calculated and presented in Fig. 7b. After 60 min photocatalytic reaction, the conversion ratios of the 2-propanol to acetone for g-CNA-1, g-CNA-2, g-CNA-3, g-CNA-4 were 18.02, 18.26, 24.87 and 14.32 ppm min−1, respectively. Blank experiments showed that no acetone was detected in the closed system in the dark, so the conversion of 2-propanol to acetone is indeed a photocatalytic process. The photocatalytic results show that porous samples exhibit much higher activities than that of sample prepared by conventional approach (4.62 ppm min−1). Enhancement in photocatalytic activity of the porous g-C3N4 can be attributed to the large specific surface area and small particle size which provides more active sites for catalysis reaction and short bulk diffusion length for reducing the recombination probability of photoexcited charge carriers.
 | ||
| Fig. 7 (a) The concentration of evolved acetone versus time and (b) evolved rate of acetone over different photocatalysts under visible light irradiation (λ > 420 nm). | ||
Generally, g-C3N4 materials have excellent thermal and chemical stability. To examine the stability of obtained porous g-C3N4, the recyclability of the as-prepared catalyst was investigated in a four-run recycling test of catalytic evolved acetone. The test was carried out over g-CNA-3, which exhibits the best photocatalytic performance. As revealed in Fig. S6,† the photocatalytic activity has not decreased significantly within four runs.
Conclusions
In summary, an artful and simple synthetic strategy aim to delay pore-forming project and reduce the carbon residues of the materials has been successfully established for the formation of a useful pore system and high performance for photocatalytic reactions. As demonstrated by the case study of g-CNA-3 synthesis, acetic acid treated melamine as precursor can inhibiting effect of the crystal growth, meanwhile, a designed “two-step” method can avoid the decomposition of pore-maker material at too early a stage. Both of these protocols can work together to formation porous g-C3N4 with negligible carbon residues. The as-obtained product have synergic advantages of porous structure, thin thickness, large specific surface area, increased band gap and high performance for photocatalytic reactions. This work represents a significant progress in scalable fabrication of porous g-C3N4, which paves the way of opening up porous g-C3N4 to broader applications for environmentally clean and provides an inspiration for the scalable production of porous polymer materials.Acknowledgements
Financial support from the education minister of Liaoning province (L2010151), National Natural Science Foundation of China [11274150, 11574124] and the Open Research Fund of Jiangsu Provincial Key Laboratory for Nanotechnology, Nanjing University. X. X. Fan is indebted to the support from the Program of Liaoning Key Laboratory of Semiconductor Light Emitting and Photocatalytic Materials.Notes and references
- J. Zhang, X. Chen, K. Takanabe, K. Maeda, K. Domen, J. D. Epping, X. Fu, M. Antonietti and X. Wang, Angew. Chem., 2010, 49, 441–444 CrossRef CAS PubMed.
- F. He, G. Chen, Y. Yu, Y. Zhou, Y. Zheng and S. Hao, Chem. Commun., 2015, 51, 425–427 RSC.
- Y. Wang, X. Wang and M. Antonietti, Angew. Chem., Int. Ed., 2012, 51, 68–89 CrossRef CAS PubMed.
- F. Raziq, C. Li, M. Humayun, Y. Qu, A. Zada, H. Yu and L. Jing, Mater. Res. Bull., 2015, 70, 494–499 CrossRef CAS.
- F. Raziq, Y. Qu, X. Zhang, M. Humayun, J. Wu, A. Zada, H. Yu, X. Sun and L. Jing, J. Phys. Chem. C, 2016, 120, 98–107 CAS.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- J. Xiao, Y. Xie, F. Nawaz, Y. Wang, P. Du and H. Cao, Appl. Catal., B, 2016, 183, 417–425 CrossRef CAS.
- S. C. Lee, H. O. Lintang and L. Yuliati, Chem.–Asian J., 2012, 7, 2139–2144 CrossRef CAS PubMed.
- F. Dong, L. Wu, Y. Sun, M. Fu, Z. Wu and S. C. Lee, J. Mater. Chem., 2011, 21, 15171 RSC.
- J. Hong, X. Xia, Y. Wang and R. Xu, J. Mater. Chem., 2012, 22, 15006 RSC.
- X. Huang, X. Qi, F. Boey and H. Zhang, Chem. Soc. Rev., 2012, 41, 666–686 RSC.
- X. Chen, J. Zhang, X. Fu, M. Antonietti and X. Wang, J. Am. Chem. Soc., 2009, 131, 11658–11659 CrossRef CAS PubMed.
- Z. Lin and X. Wang, Angew. Chem., Int. Ed., 2013, 52, 1735–1738 CrossRef CAS PubMed.
- X. Wang, X. Chen, A. Thomas, X. Fu and M. Antonietti, Adv. Mater., 2009, 21, 1609–1612 CrossRef CAS.
- S. C. Yan, Z. S. Li and Z. G. Zou, Langmuir, 2010, 26, 3894–3901 CrossRef CAS PubMed.
- J. Xue, S. Ma, Y. Zhou, Z. Zhang and M. He, ACS Appl. Mater. Interfaces, 2015, 7, 9630–9637 CAS.
- Y. Bu, Z. Chen and W. Li, Appl. Catal., B, 2014, 144, 622–630 CrossRef CAS.
- C. Han, Y. Wang, Y. Lei, B. Wang, N. Wu, Q. Shi and Q. Li, Nano Res., 2015, 8, 1199–1209 CrossRef CAS.
- X. Bai, R. Zong, C. Li, D. Liu, Y. Liu and Y. Zhu, Appl. Catal., B, 2014, 147, 82–91 CrossRef CAS.
- R. Chen, J. Zhang, Y. Wang, X. Chen, J. A. Zapien and C. S. Lee, Nanoscale, 2015, 7, 17299–17305 RSC.
- P. Niu, L. Zhang, G. Liu and H.-M. Cheng, Adv. Funct. Mater., 2012, 22, 4763–4770 CrossRef CAS.
- H. Yan, Chem. Commun., 2012, 48, 3430–3432 RSC.
- Z. Yang, A. E. Danks, J. Wang, Y. Zhang and Z. Schnepp, APL Mater., 2016, 4, 015706 CrossRef.
- X.-H. Li, J. Zhang, X. Chen, A. Fischer, A. Thomas, M. Antonietti and X. Wang, Chem. Mater., 2011, 23, 4344–4348 CrossRef CAS.
- J. Zhang, F. Guo and X. Wang, Adv. Funct. Mater., 2013, 23, 3008–3014 CrossRef CAS.
- Y. Wang, J. Zhang, X. Wang, M. Antonietti and H. Li, Angew. Chem., 2010, 49, 3356–3359 CrossRef CAS PubMed.
- Y. Zheng, L. Lin, B. Wang and X. Wang, Angew. Chem., 2015, 54, 12868–12884 CrossRef CAS PubMed.
- Y. Zhang, T. Mori and J. Ye, Sci. Adv. Mater., 2012, 4, 282–291 CrossRef CAS.
- Y. Wang, X. Wang, M. Antonietti and Y. Zhang, ChemSusChem, 2010, 3, 435–439 CrossRef CAS PubMed.
- M. Wu, J.-M. Yan, X.-w. Zhang and M. Zhao, Appl. Surf. Sci., 2015, 354, 196–200 CrossRef CAS.
- S. C. Yan, Z. S. Li and Z. G. Zou, Langmuir, 2009, 25, 10397–10401 CrossRef CAS PubMed.
- P. Yang, J. Zhao, W. Qiao, L. Li and Z. Zhu, Nanoscale, 2015, 7, 18887–18890 RSC.
- M. Groenewolt and M. Antonietti, Adv. Mater., 2005, 17, 1789–1792 CrossRef CAS.
- G. Dong and L. Zhang, J. Mater. Chem., 2012, 22, 1160–1166 RSC.
- X. Lu, K. Xu, P. Chen, K. Jia, S. Liu and C. Wu, J. Mater. Chem. A, 2014, 2, 18924–18928 CAS.
- W. Lu, T. Xu, Y. Wang, H. Hu, N. Li, X. Jiang and W. Chen, Appl. Catal., B, 2016, 180, 20–28 CrossRef CAS.
- G. Dong, W. Ho, Y. Li and L. Zhang, Appl. Catal., B, 2015, 174, 477–485 CrossRef.
- Y. Zhou, L. Zhang, W. Huang, Q. Kong, X. Fan, M. Wang and J. Shi, Carbon, 2016, 99, 111–117 CrossRef CAS.
- X. Fan, J. Gao, Y. Wang, Z. Li and Z. Zou, J. Mater. Chem., 2010, 20, 2865 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra18496k |
| This journal is © The Royal Society of Chemistry 2016 |