
Simple synthesis of novel terthiophene-based D–A1–D–A2 polymers for polymer solar cells†
Abstract
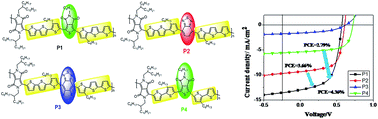
Direct arylation was used to synthesize a series of novel terthiophene (T3)-based D–A1–D–A2 polymers and D–A2–D monomers in fewer synthetic steps. In these T3-based D–A1–D–A2 polymers, pyrrolo[3,4-c]pyrrole-1,4-dione (DPP) was selected as the first acceptor A1, octyl-thieno[3,4-c]pyrrole-4,6-dione (TPD) or 2,1,3-benzothiadiazole (BT) or fluorinated benzothiadiazole (FBT) was selected as the secondary acceptor A2. T2-based polymer with the bithiophene segments (T2) as the donor was synthesized for comparison, too. UV-vis absorption, electrochemical properties, blend film morphology, and photovoltaic properties of the polymers were studied to explore the effects of the oligothiophene unit and secondary acceptor moiety (A2), meanwhile, the fluorine substitution effect was also discussed. It is shown that the change of donor segment from T2 to T3 introduces a difference in the energy levels, crystallinity, polymer:PC71BM morphology and PSC performances between the T2-based and T3-based D–A1–D–A2 polymers. Varying the secondary acceptor (A2) from BT to TPD also promotes the crystallinity and backbone planarity leading to enhanced PSC performances of the T3-based D–A1–D–A2 polymer. Although the effectiveness of fluorine substitution for tuning the UV-vis absorption, energy levels and degree of crystallinity has been demonstrated, the insufficient EDONORLUMO − EPCBMLUMO energy offset and poor miscibility of polymer:PC71BM limit the short circuit current (Jsc). In addition, the highest Jsc of 12.98 mA cm−2 is achieved for P1, while the higher HOMO level limits the open circuit voltage (Voc) and leads to a power conversion efficiency (PCE) of 4.36%.
 Please wait while we load your content...
Please wait while we load your content...

