
Solvent-controlled platinum nanocrystals with a high growth rate along 〈100〉 to 〈111〉 and enhanced electro-activity in the methanol oxidation reaction†
Abstract
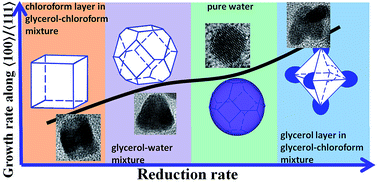
Platinum nanocrystals with different growth rates along 〈100〉 to 〈111〉 are selectively shaped using the reaction solvent, which is due to the different reduction rates in diverse reaction solutions. Electrocatalytic results show that the branched and cubic Pt/C catalysts exhibit much higher ECSA and enhanced activity of MOR, in addition, the branched Pt/C catalyst shows improved durability.
 Please wait while we load your content...
Please wait while we load your content...

