
Self-healing epoxy with a fast and stable extrinsic healing system based on BF3–amine complex†
Yi Xi Songa,
Xiao Ji Yeb,
Min Zhi Rong*c and
Ming Qiu Zhang*c
aKey Laboratory for Polymeric Composite and Functional Materials of Ministry of Education, GD HPPC Lab School of Chemistry and Chemical Engineering, Sun Yat-sen University, Guangzhou 510275, P. R. China
bGuangdong Provincial Public Laboratory of Analysis and Testing Technology, China National Analytical Center, 510070 Guangzhou, P. R. China
cMaterials Science Institute, Sun Yat-sen University, Guangzhou 510275, P. R. China. E-mail: cesrmz@mail.sysu.edu.cn; ceszmq@mail.sysu.edu.cn
First published on 10th October 2016
Abstract
To avoid the high corrosiveness of the fast-healing epoxy system previously developed by our lab, which contains a strong Lewis acid or Brønsted acid, we introduced a much less corrosive BF3–amine complex (50% boron trifluoride–2,4-dimethylaniline in 1,4-butanediol) as a replacement. The new hardener was successfully synthesized, encapsulated in silica hollow microcapsules, and embedded in an epoxy matrix with a microencapsulated cycloaliphatic epoxy monomer. Impact tests indicated that the healing system worked as expected. The self-healing epoxy composite can regain its strength within seconds at room temperature, and its self-healability remained stable after 4 months of storage. The high cationic polymerization reactivity of the cycloaliphatic epoxy accounts for the rapid restoration. It is believed that the present work represents an important step towards practical usage of the fast extrinsic self-healing strategy.
Introduction
As a next-generation technology, self-healing polymers that enable autonomous restoration of damaged structures and properties have attracted increasing research interest.1 Over time, a series of extrinsic and intrinsic healing approaches have been proposed.2 The development of new healing chemistry that can provide the greatest degree of crack rehabilitation has been the major focus of this research. Comparatively, only a few studies have focused on healing speed, and much less attention has been paid to the fast recovery of mechanical strength.3–12In fact, cracks propagate very rapidly in solid materials.13,14 Cracks should be eliminated soon after initiation to prevent their growth and the resulting instability of the materials. In this context, self-healing speed is of equal importance to healing efficiency. Our previous exploration in this direction indicated that crack healing of damaged epoxy can be completed within 20 to 100 s when microencapsulated instant hardener (antimony pentafluoride8,9 or trifluoromethanesulfonic acid10) is used in cooperation with epoxy monomer capsules; this is due to the rapid curing of the released epoxy, catalyzed by the hardener. This time is much shorter than those required by other systems (hours or dozens of minutes).
However, it is worth noting that antimony pentafluoride and trifluoromethanesulfonic acid are very strong Lewis and Brønsted acids, respectively; these are difficult to handle during the manufacture and large-scale production of microcapsules. To overcome this disadvantage while still achieving fast healing for practical applications, we began to seek a stable, safe, easy-processing hardener and a matching epoxy as the components of a new healing agent. After many attempts, we finally found that 50% boron trifluoride–2,4-dimethylaniline complex (BF3–DMA) in 1,4-butanediol is a promising candidate which can cure a cycloaliphatic epoxy, 3,4-epoxycyclohexylmethyl-3,4-epoxycyclohexanecarboxylate (trade name: Araldite® CY179), within dozens of seconds at room temperature. This curing speed is much faster than that of the (C2H5)2O·BF3–epoxy pair,15,16 another rapidly solidifying system containing BF3. BF3–DMA complex is a solid, which ensures its stability and low moisture absorbance. The solution of BF3–DMA in 1,4-butanediol (BF3–DMA/BDO) is much less corrosive than the aforementioned antimony pentafluoride and trifluoromethanesulfonic acid, and it can be readily mixed with epoxy.
In the mid-1950's, the Shell Chemical Co. disclosed a boron trifluoride (BF3) monoethylamine adduct for curing epoxy resin.17 The reactivity or dissociation of the BF3–amine adduct is dependent on the relative basicity of the amine with respect to the acidic BF3. Moreover, it has been demonstrated that aromatic primary amines, which are much weaker bases than aliphatic primary amines, in the form of complexes can cure epoxides at much lower temperatures than aliphatic amine complexes.18 On the other hand, when the protons of an electrophilic reagent attack oxygen atoms during the cationic additive polymerization reaction, cycloaliphatic epoxy is more reactive than oxirane because the epoxy groups of the former possess a higher electron cloud density than those of the latter. This may explain the fast curing of cycloaliphatic epoxy in the presence of BF3–DMA/BDO.
Cycloaliphatic epoxy is a unique class of epoxy containing non-aromatic saturated rings; it possesses notable features, such as inherently low viscosity and excellent weathering and electrical performances. As a member of the cycloaliphatic epoxy family, CY179 has found extensive commercial applications. Formulations incorporating acid anhydride as a curing agent in this resin provide cured products with high glass transition temperatures in the range of 200 °C as well as high strength retention at elevated temperatures. Lewis acid catalysts can also be used as curing agents that operate after cationic polymerization.
Herein, BF3–DMA/BDO and cycloaliphatic epoxy are respectively encapsulated and embedded in an epoxy matrix to produce a self-healing epoxy based on a dual microcapsules approach.2 The curing mechanism, healing performance and influential factors of the system are investigated with the aim of providing a user-friendly method to replace earlier versions. The outcome presented herein will promote the self-healing technique and increase its viability for practical engineering applications.
Experimental
Materials and reagents
The diglycidyl ether of bisphenol A (trade name EPON 828), an epoxy, was purchased from Shell Corporation to be used as the matrix of the composite. Cycloaliphatic epoxy (trade name: Araldite CY179), the polymerizable component of the healing agent, was purchased from Huntsman Corporation. 50% boron trifluoride–2,4-dimethylaniline complex in 1,4-butanediol (BF3–DMA/BDO), the hardener component of both the matrix and the healing agent, was synthesized in our laboratory.Ethanol, tetraethyl orthosilicate (TEOS), polyvinylpyrrolidone (PVP), hexadecyltrimethylammonium bromide (CTAB), styrene and methyl methacrylate were purchased from Aladdin Reagent Co., Shanghai, China. Prior to use, styrene and methyl methacrylate were washed with sodium hydroxide aqueous solution (5%) and washed three times with water, then dried over anhydrous magnesium sulphate. 2,2-Azobisisobutyronitrile (AIBN) was obtained from Sigma-Aldrich and purified by recrystallization. Ammonia (38 wt%) and dichloromethane (dried with 4 Å molecular sieves) were purchased from Guangzhou Chemical Reagent Factory, China. Melamine and formaldehyde were provided by Shanghai Medical Group Reagent Co., China.
Preparation of BF3–DMA/BDO
The BF3–amine adducts were generally acquired by an interchange reaction of BF3 etherate with amine in solvent. In this work, 121.18 g (1 mol) 2,4-dimethylaniline was firstly dissolved in 500 mL methylene chloride (dried with molecular sieves), and then 150 g (1.05 mol) boron trifluoride diethyl ether was dropwise added at a speed of 5 s per d below −10 °C. Afterwards, the solution was maintained at room temperature for 12 h under stirring. The reaction product, i.e. BF3–DMA, was obtained in the form of a light yellow powder by filtering, washing, and vacuum drying at 40 °C. The structure of the product was confirmed by Fourier transform infrared (FTIR) spectra, nuclear magnetic resonance (NMR) spectra and the mass spectrum (Fig. S1–S4, ESI†). Finally, BF3–DMA was dissolved in 1,4-butanediol at a weight ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, yielding the hardener BF3–DMA/BDO.
:1, yielding the hardener BF3–DMA/BDO.
Preparation of silica walled microcapsules containing BF3–DMA/BDO
Due to the high reactivity of BF3–DMA/BDO, conventional encapsulation techniques such as in situ polymerization in an oil-in-water emulsion19 are not suitable for use. Instead, we first prepared hollow silica microcapsules and then infiltrated the hardener into the hollow silica microcapsules. The main procedures for preparing this type of microcapsule have been reported elsewhere.20 Firstly, monodispersed poly(styrene–methylmethacrylate) (PS–MMA) particles (average diameter: 5 μm) were prepared by dispersion polymerization. Then, the precursor TEOS was coated onto the PS–MMA particles by the sol–gel method. Typically, 5 g PS–MMA emulsion (containing 2 g PS–MMA particles), 0.25 g CTAB, 20 mL deionized water and 50 mL ethanol were added to a 250 mL flask with ultrasonication for 15 min, followed by mechanical stirring at 70 °C for 30 min. Afterwards, 1 g TEOS in 10 mL ethanol solution was added dropwise, and the mixture was reacted for an additional 8 h. When the PS–MMA template was removed by high temperature calcination at 550 °C, hollow silica microcapsules were obtained.Because the fluidic hardener will be infiltrated into the hollow silica microcapsules, the pore size of the capsule shell plays an important role. The size must be appropriate to facilitate vacuum-aided uptake while hindering outflow of the infiltrated hardener. In this context, the pore size of the shell of the hollow silica microcapsules is accurately controlled by the amount of CTAB (Fig. 1). During the preparation of the hollow silica microcapsules through a sol–gel route in a mixture of ethanol and water, the added surfactant CTAB self-assembles into rod-like micelles which act as a template and induce assembly of TEOS. In addition, CTAB plays a key role to control the pore size when it is removed in the course of high temperature calcination. When the CTAB content is appropriate, thermal decomposition of CTAB in the process of calcination leads to the formation of nano-sized worm-like pores on the walls of the hollow silica microcapsules (Fig. 1a and b). When excessive CTAB is added, many more micelles self-assemble; therefore, an increased number of pores with larger sizes remain after calcination (Fig. 1c). In extreme situations, micron-sized voids or even wall collapse are observed (Fig. 1d).
 | ||
| Fig. 1 Scanning electron microscopic (SEM) photos of silica hollow capsules prepared with different contents of CTAB (in terms of the weight ratio of CTAB/TEOS/PS–MMA): (a and b) 1/4/8, (c) 1/2/4, and (d) 3/4/8. | ||
By a vacuum-aided process,21 the hardener BF3–DMA/BDO was infiltrated into the hollow silica microcapsules to produce the hardener-loaded microcapsules. Typically, 0.5 g hollow silica microcapsules (CTAB/TEOS/PS–MMA = 1/4/8) were submerged in the hardener. After sealing and heating to 50 °C, the system was evacuated to a vacuum degree of 0.001 to 0.002 mbar. After 60 h, the resulting hardener-loaded microcapsules were filtered and purged with dichloromethane. The core content was found to be about 50 wt% as measured by thermogravimetric analysis (TGA, Fig. S5, ESI†). Additionally, the TGA curves of other hardener-loaded microcapsules prepared with different CTAB contents were also given to show the influence of pore size on the core content (Fig. S6, ESI†).
Preparation of epoxy-loaded microcapsules and self-healing epoxy composite
Cycloaliphatic epoxy monomer (CY179) was encapsulated by poly(melamine formaldehyde) (PMF) as mentioned in ref. 22. Compared to the hardener-loaded microcapsules, the epoxy capsules are more easily synthesized. Their average diameter, wall thickness and core content are 60 μm, 1.7 μm and 88 wt%, respectively, as determined by SEM (Fig. 2) and the extraction method.22 Additionally, Fourier transform infrared (FTIR) spectra of the epoxy-loaded microcapsules and their core and wall substances reveal that all the characteristic peaks of epoxy and PMF are observed in the spectrum of the epoxy microcapsules (Fig. S7, ESI†). This provides one piece of evidence for the successful preparation of the capsules. | ||
| Fig. 2 SEM photos of (a and b) epoxy (CY179)-loaded microcapsules and (c and d) the crushed microcapsules, showing the wall thickness. | ||
Unfilled epoxy specimens were cast from a mixture of 10 parts hardener (BF3–DMA/BDO) and 100 parts epoxy (EPON 828). The self-healing epoxy specimens were prepared by uniformly blending 15 wt% epoxy (CY179)-loaded microcapsules and 2 wt% hardener-loaded microcapsules with the above mixture of hardener and epoxy (EPON 828). After degasification, the compounds were poured into a preheated silicone rubber mold and successively cured at 60 °C for 12 h, 80 °C for 12 h and 100 °C for 1 h. Dynamic mechanical analysis (DMA), shown in Fig. S8, ESI,† shows that the epoxy (EPON 828) was fully cured under the present curing conditions, as no additional peak appears during the cyclic tests. Moreover, the 3D X-ray image (Fig. S9, ESI†) confirms that the healing capsules are sufficiently robust to survive compounding.
Characterization
FTIR spectra were collected by a Thermo-Nicolet Nexus 670 spectroscope. 1H-NMR and 19F-NMR spectra were measured using a Bruker Avance III spectrometer (400 MHz). The high resolution mass spectrum was recorded with a Thermo MAT95XP (ESI) mass spectrometer.Morphological observation was conducted on a HITACHI model S-4800 field emission scanning electron microscope (SEM). The spatial arrangement of the microcapsules in the composite was inspected using a nanoVoxel-2100 X-ray 3D microscope (Sanying Precision Instruments Co. Ltd., China).
Differential scanning calorimetry (DSC) was performed on a TA Instruments Q10 instrument at a heating rate of 10 °C min−1 with nitrogen purge. Thermogravimetric analysis (TGA) was performed on a TA Instruments TGA Q 50 at a heating rate of 10 °C min−1 with nitrogen purge. Dynamic mechanical analysis (DMA) was carried out with a 01Db-metravib-DMA25 apparatus using single cantilever mode under 3 Hz at a heating rate of 2 °C min−1 in nitrogen.
To monitor the exothermic properties of the curing reaction, optical pyrometry23 was used. Different weight percentages (1, 5, 10 and 15%) of the hardener (50% boron trifluoride–2,4-dimethylaniline complex in 1,4-butanediol) were injected into epoxy monomer (CY179, 2 g) under stirring, respectively. The temperature change of the system was recorded by means of a thermal camera (VarioCAM hr head780, InfraTec GmbH, Germany).
The healing capability of the epoxy composite damaged by monotonic fractures was assessed by impact test.24 The test was conducted at 25 °C on an Izod notched specimen (52.8 × 12.3 × 10.1 mm3) according to ASTM D265-034 using a JJ-20 impact tester produced by Changchun Research Institute for Testing Machines Co. Ltd., China. The specimen was impacted to failure, and then the cracked faces were allowed to come back into contact as fast as possible. The specimen was healed at 25 °C under a gentle pressure of ∼0.2 MPa for a period of time. Finally, the healed specimen was impacted again. The healing efficiency was calculated from the ratio of the impact strength of the healed and virgin materials. Each batch included five specimens to yield averaged values.
Results and discussion
Mechanism of the reaction between BF3–DMA/BDO and cycloaliphatic epoxy
Fig. 3 shows the temperature increase induced by the curing reaction of cycloaliphatic epoxy (CY179) catalyzed by the hardener (BF3–DMA/BDO). Soon after the hardener is mixed with the epoxy, rapid polymerization occurs, and the system temperature increases above room temperature within a few seconds. When surveying the effect of the hardener dosage, it was found that the time to reach the maximum temperature was shortest at 5 to 10 wt%. This is because of the cationic chain polymerization mechanism. That is, the curing speed increases with increasing catalyst concentration below a minimum threshold. As long as the catalyst amount exceeds the critical value, the curing speed is nearly independent of the catalyst content. The longer time required by the system with 15 wt% hardener may be due to the reduced uniformity of the mixture. The contact between the epoxy monomer and the hardener is greatly reduced by the curing of the epoxy at the early stage of the reaction. On the other hand, the highly exothermic property of the curing implies that this reaction can occur in a cold environment without external heating. Sub-ambient temperature self-healing would thus be possible.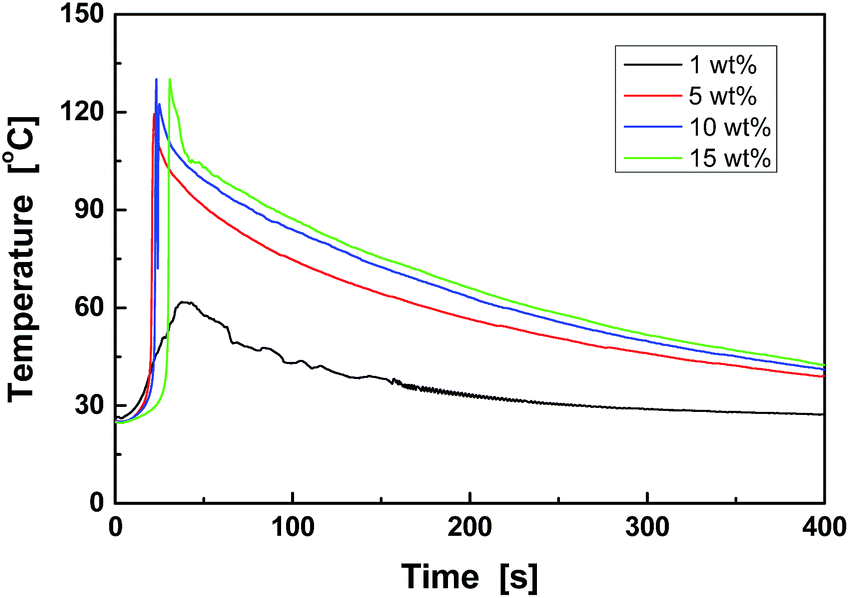 | ||
| Fig. 3 Time dependence of the temperature of epoxy (CY179) cured by different weight percentages of hardener (BF3–DMA/BDO). | ||
To understand whether the reactivity of the hardener changes after encapsulation, 0.5 g cycloaliphatic epoxy (CY179) and 0.1 g hardener-loaded microcapsules were mixed, ground and monitored by thermal camera. As shown in Fig. 4a, the system temperature increases drastically due to the rapid exothermic property of the curing. These results manifest that the encapsulated hardener can catalyze fast curing of the epoxy, as expected, which ensures the performance of the healing system embedded in the epoxy composite. In contrast, the reaction between BF3–DMA/BDO and the oxirane of the diglycidyl ether of bisphenol A (EPON 828) is much slower; therefore, the exothermic behavior of the curing can be monitored by DSC (Fig. 4b. Note: DSC is not applicable to inspect the curing of the cycloaliphatic epoxy (CY179) with BF3–DMA/BDO because polymerization proceeds significantly before the components are evenly mixed).
 | ||
| Fig. 4 (a) Temperature profile of the ground mixture of epoxy (CY179) and hardener-loaded microcapsules (5/1 by weight). (b) DSC heating traces of the mixtures of (curve 1) epoxy (EPON 828) and hardener (BF3–DMA/BDO, 10/1 by weight), and (curve 2) epoxy (EPON 828) and crushed hardener-loaded microcapsules. Rate of heating for DSC measurements: 10 °C min−1. | ||

In the presence of a relatively high concentration of alcohol, because alcohol is more nucleophilic than the epoxy monomer, the oxonium ion is prone to react first with the alcohol; then, the product reacts with the epoxy monomer.

The remaining alcohol in the system can react in the same way.
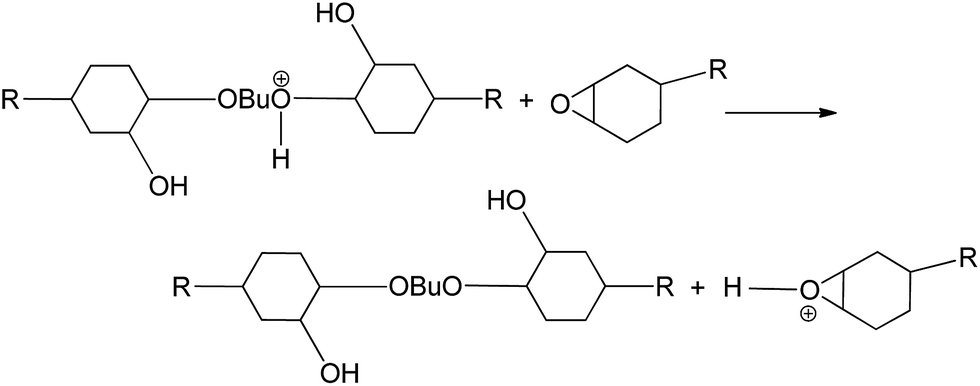
To date, the curing mechanism of epoxy catalyzed by amine–boron trifluoride complex has been controversial. Firstly, it was believed that the epoxy is cured by the BF3 liberated by thermal dissociation of the complex.25 Later, another mechanism was proposed which postulated that after heat treatment, BF3–amine dissociates to a tetrafluoroborate salt.26 The latter reacts with the epoxy as the true catalyst. Similarly, Smith et al.27–29 suggested that at temperatures above 80 °C, the BF3 complex is rapidly hydrolyzed to HBF4 and/or other hydrolysates, which catalyzes the curing of the epoxy. Bouillon30 considered that a proton is released during thermal dissociation of the complex followed by an addition reaction of this proton to the oxygen atom of the oxirane ring, yielding an oxonium ion. The above analyses claim that the true catalysis mechanism of the epoxy involves thermal dissociation of the amine–boron trifluoride complex; however, this is inapplicable to the present system, as curing of the epoxy catalyzed by BF3–DMA/BDO occurs at room temperature without heat pre-treatment.
Meanwhile, Harris and Temin18 proposed that a proton is contributed to the oxygen of the epoxy group from the amine–boron trifluoride complex, creating a partial positive charge. This consideration is supported by studies of the curing reaction of epoxy with BF3 complex using nuclear magnetic resonance, infrared and mass spectroscopy.31 Additionally, some researchers30,32–35 showed that in the presence of a relatively large concentration of alcohol groups, the mechanism of polymerization switches from the activated chain end mechanism to the activated monomer mechanism. Based on these pioneering efforts, we deduce that the polymerization mechanism for cycloaliphatic epoxy cured with BF3–DMA/BDO follows the cationic polymerization mechanism, as shown in Fig. 5.
 | ||
| Fig. 5 Initiation and propagation of the cationic polymerization of cycloaliphatic epoxy catalyzed by BF3–DMA/BDO. | ||
As shown in Fig. 4, cycloaliphatic epoxy possesses much higher cationic polymerization reactivity than the diglycidyl ether of bisphenol A. Similar phenomena have been reported by other groups;36,37 however, the underlying mechanism has not been clearly revealed. It was suggested that the cycloaliphatic epoxy group has higher electronic density on its oxygen atom and thus is more labile to cationic polymerization.38 Moreover, the rigidity and steric bulk of the cycloaliphatic ring cannot easily adopt the planar cyclic conformation necessary for the formation of hydrogen-bonded structures,39 which may additionally contribute to the high reactivity exhibited by cycloaliphatic epoxides in cationic ring-opening polymerizations conducted at room temperature and lower temperatures. However, neither experimental nor theoretic data exist to support these hypotheses.
To explain the fast curing behavior of cycloaliphatic epoxy catalyzed by BF3–DMA/BDO observed in this work, a theoretical simulation was carried out using Chem3D software to calculate the charge density of the oxonium ion in the cycloaliphatic epoxy molecule. The chemical structures of the epoxy resin (CY179 and EPON 828) molecules were drawn using ChemDraw Ultra 12.0 and then exported to ChemBio3D Ultra 14.0. After structure optimization via MM2 minimize energy processing, the total energy of each molecule was determined. Meanwhile, the charge density of each oxonium ion was estimated by the extended Huckel method. The total energy of epoxy CY179 with an oxonium ion (283.3 kcal mol−1) is higher than that of epoxy EPON 828 with an oxonium ion (262.0 kcal mol−1), meaning that epoxy CY179 is more reactive. Furthermore, the positive charge value of the epoxy CY179 oxonium ion (about 0.33) is smaller than that of the epoxy EPON 828 oxonium ion (approximately 0.35), which indicates that the electron density of the former is higher than that of the latter. For an oxonium ion, a higher electron density corresponds to higher reactivity. As a result, the oxonium ion of epoxy CY179 should be more reactive during cationic polymerization.
Evaluation of self-healing performance
Because the above pilot studies verified the suitability of the dual microcapsules system for fast curing, its performance in an authentic epoxy composite was evaluated by impact test, as described in the following.As shown in Fig. 6, healing proceeds quite rapidly at 25 °C, meaning that (i) the healing agent can be liberated from the capsules upon cracking of the composite, and (ii) curing of the released healing agent is completed within a short time without manual intervention. The first healing efficiency of 32.5% was measured at 40 s. With increasing time, the healing efficiency increases up to 600 s and then reaches equilibrium. In other words, the maximum healing efficiency (70.3%) is achieved at 600 s. Although this time is slightly longer than that required by healing systems with antimony pentafluoride8,9 or trifluoromethanesulfonic acid10 using the same test protocol (i.e. 100 s), it is much shorter than that of other healing agent systems based on cationic polymerization (such as the (C2H5)2O·BF3–epoxy pair, 2 h (ref. 15 and 16)).
 | ||
| Fig. 6 Dependences of the Izod impact strength and healing efficiency of healed self-healing epoxy composites on healing time at 25 °C. The epoxy- and hardener-loaded capsule contents are 15 and 2 wt%, respectively. | ||
The morphology of the fractured surfaces of the healed composite (Fig. 7) shows numerous lumps of cured resin and uneven membrane from the crosslinked healing agent. This demonstrates that the fluidic healing agent burst from the broken microcapsules, spread over the fractured surface and rejoined the split specimen at room temperature.
 | ||
| Fig. 7 SEM images of the impact fracture surface of the healed self-healing epoxy composite. The contents of epoxy- and hardener-loaded capsules are 15 and 2 wt%, respectively. The specimen was fractured by the first impact test, healed at 25 °C for 600 s, and then fractured by the second impact test. Photos (b–d) are magnified views of different parts of (a). | ||
Durability is an important factor for effective usage of materials. We measured the healing efficiency of self-healing epoxy composites that were stored at room temperature for different periods of time. The data in Fig. 8 manifest that the healing capability does not decay within 4 months. Clearly, there is no leakage of healing agent from the capsules in the manufactured composite, which provides long-term stability for the healing system. Otherwise, a continuous deterioration of healing efficiency would be perceived.40
 | ||
| Fig. 8 Healing efficiency of the self-healing epoxy composites as a function of storage time at room temperature. The epoxy- and hardener-loaded capsule contents are 15 and 2 wt%, respectively. Healing was conducted at 25 °C for 600 s. | ||
In addition to healing time, which is the main concern of this work, the effects of the loading of healing capsules should also be understood. As demonstrated by optical pyrometry in Fig. 3, 5 to 10 wt% hardener is sufficient for curing the cycloaliphatic epoxy (CY179). When the healing agent is encapsulated and embedded in the epoxy matrix, however, the amounts of epoxy and hardener released from the ruptured capsules at the cracked plane are rarely predicted. For this reason, a series of epoxy composites filled with different proportions of epoxy-loaded microcapsules and hardener-loaded microcapsules were prepared and tested to optimize the synthesis. As illustrated in Fig. 9, when 5 wt% epoxy-loaded microcapsules were added, the healing efficiency of the composite (∼52%) remained nearly unchanged even though the content of the hardener-loaded capsules increased from 1 to 5 wt%. This is likely due to insufficient coverage of the healing membrane on the fractured surface. In the case of the 10 wt% epoxy-loaded microcapsules, the hardener-loaded microcapsules were insufficient at first; however, the critical threshold was soon reached if the content was 2 wt% or higher. Nevertheless, the healing efficiency (∼65%) still suffers due to a shortage of the released healing agent. Only when the content of the epoxy-loaded microcapsules is 15 or 20 wt% and that of the hardener capsules is 2 to 5 wt%, the epoxy/hardener ratio exceeds the threshold and the released healing agent is sufficient to cover the entire fracture surface. As a result, the measured healing efficiency reached the highest value (∼70%) and became independent of the capsule contents.
 | ||
| Fig. 9 Dependences of the healing efficiency of the self-healing epoxy composites on the contents of (a) hardener-loaded microcapsules with constant content of epoxy (CY179)-loaded microcapsules and (b) epoxy (CY179)-loaded microcapsules with constant content of hardener-loaded microcapsules. Healing was conducted at 25 °C for 600 s. | ||
In general, the incorporation of a microencapsulated healing agent into a polymer leads to a reduction in mechanical properties.41,42 This is acceptable, due to the acquisition of the additional functionality of self-healability; however, the decrease in the properties should be controlled to a low level. The influence of different contents of the hardener- and epoxy (CY179)-loaded microcapsules on the impact strength is depicted in Fig. 10. Because the diameter of the hardener-loaded microcapsules (5 μm) is much smaller than that of the epoxy-loaded microcapsules (average diameter: 60 μm), the impact strength of the composites containing the hardener microcapsules slightly decreases compared with the composites containing the epoxy microcapsules within the same range of capsule contents (0 to 5 wt%, for example). The difference implies that the included microcapsules serve as stress concentrative points in the composites.
 | ||
| Fig. 10 Dependences of the Izod impact strength of the epoxy composites on the contents of (a) hardener-loaded microcapsules and (b) epoxy (CY179)-loaded microcapsules. | ||
Fig. 11 further reflects the situations of authentic self-healing epoxy composites containing both epoxy- and hardener-loaded microcapsules. The collective behavior of the larger epoxy capsules and smaller hardener capsules is dominated by the larger capsules. Taking the results shown in Fig. 9 and 11 into account, a formulation with 15 wt% epoxy capsules and 2 wt% hardener capsules is believed to be optimal to offer the highest healing efficiency.
 | ||
| Fig. 11 Dependences of the Izod impact strength of the self-healing epoxy composites on the contents of (a) hardener-loaded microcapsules with constant content of epoxy (CY179)-loaded microcapsules and (b) epoxy (CY179)-loaded microcapsules with constant content of the hardener-loaded microcapsules. | ||
Conclusions
To conclude, 50% boron trifluoride–2,4-dimethylaniline complex in 1,4-butanediol was successfully synthesized and included in hollow silica microcapsules; this formed a fast healing system with microencapsulated cycloaliphatic epoxy monomers. The rapid curing of the cycloaliphatic epoxy catalyzed by the BF3–amine complex was explained by the higher cationic polymerization reactivity of the cycloaliphatic epoxy originating from the higher electron density of its oxonium ions. The epoxy composites embedded with these two types of microcapsules can be healed within seconds at room temperature, as characterized by the restoration of impact strength. The healing speed is comparable to those of healing systems incorporating antimony pentafluoride/epoxy8,9 and trifluoromethanesulfonic acid/epoxy10 pairs. More importantly, the present fast curing agent is much more stable, safer and more easily produced, which is conducive to practical applications.The disadvantage of the healing agent lies in the fact that the high reaction speed results in incomplete curing of the released epoxy monomer; as a result, the maximum healing efficiency observed above was less than 100%. Decreasing the viscosity and increasing the flowability of the hardener, which promotes mixing of the components of the healing agent, may help to increase the reaction degree. Additionally, the introduction of a second hardener that leads to “post-curing” may be a solution. The feasibility of these plans is being examined in our lab.
Acknowledgements
The authors are grateful for the support from the Natural Science Foundation of China (Grants: 51273214, 51673219 and 51333008), the Natural Science Foundation of Guangdong (Grants: 2010B010800021 and S2013020013029), and the Science and Technology Program of Guangzhou (Grants: 2014J4100121 and 201604010105).Notes and references
- M. Q. Zhang and M. Z. Rong, Self-Healing Polymers and Polymer Composites, John Wiley & Sons, Inc., Hoboken, 2011 Search PubMed.
- D. Y. Zhu, M. Z. Rong and M. Q. Zhang, Prog. Polym. Sci., 2015, 49–50, 175 CrossRef CAS.
- S. Neumann, D. Dohler, D. Strohl and W. H. Binder, Polym. Chem., 2016, 7, 2342 RSC.
- M. Vatankhah-Varnoosfaderani, S. Hashmi, A. GhavamiNehad and F. J. Stadler, Polym. Chem., 2014, 5, 512 RSC.
- C. Wang, N. Liu, R. Allen, J. B. H. Tok, Y. P. Wu, F. Zhang, Y. S. Chen and Z. N. Bao, Adv. Mater., 2013, 25, 5785 CrossRef CAS PubMed.
- E. Z. Zhang, T. Wang, L. Zhao, W. X. Sun, X. X. Liu and Z. Tong, ACS Appl. Mater. Interfaces, 2014, 6, 22855 CAS.
- Y. Li, B. Ge, X. H. Men, Z. Z. Zhang and Q. J. Xue, Compos. Sci. Technol., 2016, 125, 55 CrossRef CAS.
- X. J. Ye, J. L. Zhang, Y. Zhu, M. Z. Rong, M. Q. Zhang, Y. X. Song and H. X. Zhang, ACS Appl. Mater. Interfaces, 2014, 6, 3661 CAS.
- (a) X. J. Ye, Y. Zhu, Y. C. Yuan, Y. X. Song, G. C. Yang, M. Z. Rong and M. Q. Zhang, eXPRESS Polym. Lett., 2015, 9, 177 CrossRef CAS; (b) P. Vijayan and M. A. AlMaadeed, eXPRESS Polym. Lett., 2016, 10, 506 CrossRef.
- (a) X. J. Ye, Y. X. Song, Y. Zhu, G. C. Yang, M. Z. Rong and M. Q. Zhang, Compos. Sci. Technol., 2014, 104, 40 CrossRef CAS; (b) C. M. Li, J. J. Tan, J. W. Gu, L. Qiao, B. L. Zhang and Q. Y. Zhang, Compos. Sci. Technol., 2016, 123, 250 CrossRef CAS; (c) H. Zhang, P. Wang and J. Yang, Compos. Sci. Technol., 2014, 94, 23 CrossRef CAS.
- M. Q. Zhang, eXPRESS Polym. Lett., 2015, 9, 84 CrossRef.
- W. M. Xu, M. Z. Rong and M. Q. Zhang, J. Mater. Chem. A, 2016, 4, 10683 CAS.
- A. J. Rosakis, O. Samudrala, R. P. Singh and A. Shukla, J. Mech. Phys. Solids, 1998, 46, 1789 CrossRef CAS.
- K. B. Broberg, Int. J. Fract., 1989, 39, 1 CrossRef.
- D. S. Xiao, Y. C. Yuan, M. Z. Rong and M. Q. Zhang, Adv. Funct. Mater., 2009, 19, 2289 CrossRef CAS.
- (a) D. S. Xiao, Y. C. Yuan, M. Z. Rong and M. Q. Zhang, Polymer, 2009, 50, 2967 CrossRef CAS; (b) D. S. Xiao, M. Z. Rong and M. Q. Zhang, Polymer, 2007, 48, 4765 CrossRef CAS.
- C. H. Smith, Mod. Plast., 1969, 46, 118 CAS.
- J. J. Harris and S. C. Temin, J. Appl. Polym. Sci., 1966, 10, 523 CrossRef CAS.
- Y. C. Yuan, M. Z. Rong and M. Q. Zhang, Polymer, 2008, 49, 2531 CrossRef CAS.
- X. J. Ye, Z. X. Ma, Y. X. Song, J. J. Huang, M. Z. Rong and M. Q. Zhang, Chin. Chem. Lett., 2014, 25, 1565 CrossRef CAS.
- D. S. Xiao, Y. C. Yuan, M. Z. Rong and M. Q. Zhang, Polymer, 2009, 50, 560 CrossRef CAS.
- Y. C. Yuan, M. Z. Rong, M. Q. Zhang, J. Chen, G. C. Yang and X. M. Li, Macromolecules, 2008, 41, 5197 CrossRef CAS.
- J. V. Crivello, J. Mater. Chem. A, 2009, 47, 5639 CAS.
- S. A. Hayes, W. Zhang, M. Branthwaite and F. R. Jones, J. R. Soc., Interface, 2007, 4, 381 CrossRef CAS PubMed.
- M. Tackie and G. C. Martin, J. Appl. Polym. Sci., 1993, 48, 793 CrossRef CAS.
- J. A. Happe, R. J. Morgan and C. M. Walkup, Polymer, 1985, 26, 827 CrossRef CAS.
- R. E. Smith, F. N. Larsen and C. L. Long, J. Appl. Polym. Sci., 1984, 29, 3697 CrossRef CAS.
- R. E. Smith, F. N. Larsen and C. L. Long, J. Appl. Polym. Sci., 1984, 29, 3713 CrossRef CAS.
- R. E. Smith and C. H. Smith, J. Appl. Polym. Sci., 1986, 31, 929 CrossRef CAS.
- N. Bouillon, J. P. Pascault and L. Tighzert, Makromol. Chem., 1990, 191, 1403 CrossRef CAS.
- C. S. Chen and E. M. Pearce, J. Appl. Polym. Sci., 1989, 37, 1105 CrossRef CAS.
- P. Kubisa, Makromol. Chem., Macromol. Symp., 1988, 13, 203 CrossRef.
- N. Bouillon, J. P. Pascault and L. Tighzert, Makromol. Chem., 1990, 191, 1417 CrossRef CAS.
- N. Bouillon, J. P. Pascault and L. Tighzert, Makromol. Chem., 1990, 191, 1435 CrossRef CAS.
- T. Vidil and F. Tournihac, Macromolecules, 2013, 46, 9240 CrossRef CAS.
- Y. Zhou, N. Tran, Y. C. Lin, Y. Z. He and F. G. Shi, IEEE Trans. Adv. Packag., 2008, 31, 484 CrossRef CAS.
- Y. Morita, J. Appl. Polym. Sci., 2005, 97, 1395 CrossRef CAS.
- K. Morio, H. Murase and H. Tsuchiya, J. Appl. Polym. Sci., 1986, 32, 5727 CrossRef CAS.
- U. Bulut and J. V. Crivello, Macromolecules, 2005, 38, 3584 CrossRef CAS.
- T. Yin, M. Z. Rong, M. Q. Zhang and J. Q. Zhao, Smart Mater. Struct., 2009, 18, 074001 CrossRef.
- D. Y. Zhu, G. S. Cao, W. L. Qiu, M. Z. Rong and M. Q. Zhang, Polymer, 2015, 69, 1 CrossRef CAS.
- D. Y. Zhu, J. W. Guo, G. S. Cao, W. L. Qiu, M. Z. Rong and M. Q. Zhang, J. Mater. Chem. A, 2015, 3, 1858 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Fourier transform infrared (FTIR) spectra, nuclear magnetic resonance (NMR) spectra, mass spectrum, pyrolytic behaviors, temperature dependences of tanδ, and X-ray 3D microscopic image. See DOI: 10.1039/c6ra17976b |
| This journal is © The Royal Society of Chemistry 2016 |