
Synthesis and characterization of functionalized titania-supported Pd catalyst deriving from new orthopalladated complex of benzophenone imine: catalytic activity in the copper-free Sonogashira cross-coupling reactions at low palladium loadings
Kazem Karami*a,
Sedigheh Abedanzadeha and
Pablo Hervésb
aDepartment of Chemistry, Isfahan University of Technology, Isfahan 84156/83111, Iran. E-mail: karami@cc.iut.ac.ir
bDepartamento de Quimica Fisica, Universidade de Vigo, 36310 Vigo, Spain. E-mail: jherves@uvigo.es
First published on 26th September 2016
Abstract
The present work describes the preparation of organically modified TiO2-supported Pd catalyst originated from the new benzophenone imine-derived CN-palladacycle. The heterogeneous organic–inorganic hybrid catalyst system has been characterized by FT-IR, XRD, SEM, EDX, TEM and XPS techniques and exhibited good catalytic activity in the Sonogashira cross-coupling reactions of phenylacetylene with aryl halides under copper-, amine- and phosphine-free conditions, in conjunction with the ultra low catalyst Pd-loading. Significantly, the heterogeneous Pd catalyst allowed the reaction of phenylacetylene with aryl iodides to improve in excellent yields under very mild conditions using green solvent. Finally, the reusability of the supported Pd-complex was investigated by multiple reuses of the supported catalyst in subsequent Sonogashira cross-couplings.
1. Introduction
Nitrogen-containing palladacycles have received growing interest due to their applications in many areas including organic synthesis, optical resolution, design of new metallomesogens and antitumoral drugs, asymmetric synthesis, intermolecular aromatic C–H bond activation, synthesis and reactivity of organometallic complexes with biologically important ligands and drug delivery.1–10 They show promising catalytic activities as a new family of homogeneous and heterogeneous palladium catalyst precursors,11–15 due to the fact that N-donor ligands strongly donate electrons to the metal center, stabilizing various oxidation states of metals and subsequently influencing their reactivity.16,17The Sonogashira coupling reaction of terminal alkynes with aryl halides is one of the most important methods which has been widely applied for the synthesis of natural products, pharmaceutical compounds and polymeric materials.18–23 This reaction is generally catalyzed by homogeneous Pd complexes in the presence of toxic phosphine ligands, using copper salts as co-catalysts and amine as a solvent or base, under inert condition which has so many drawbacks.24–29 Consequently, there have been many efforts to introduce highly active and easily reusable catalysts through convenient Cu-, amine and phosphine-free methods under environmentally friendly conditions.
Supported palladium catalysts with high dispersion and narrow size distribution of palladium particles would address the limited practical application of homogeneous palladium catalysts and exhibit high activity, recyclability, low palladium loading and promising performances in the synthetic and industrial chemistry. In this regard, a wide variety of solid materials as heterogeneous Pd supports including active carbon,30 mesoporous silica,31 metal oxides,32 zeolites,33 magnetic materials,34 hydrotalcites,35 hydroxyapatite,36 and organic–inorganic hybrid37 have been investigated. However, many heterogeneous catalytic systems suffer from high palladium loading and/or low catalytic activities due to the palladium agglomeration and catalyst deactivation.38
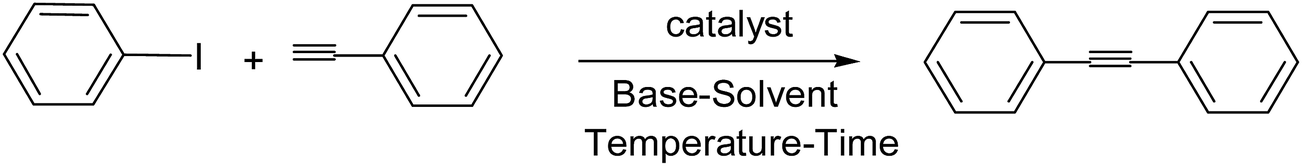
In this study, we report the synthesis and characterizations of a nitrogen-containing orthopalladated complex driving from bis[4-(dimethylamino)phenyl]methaniminium chloride (Auramine-O). In continuation of our interest in developing new supported catalysts,39–42 herein we use a strategy to convert a homogeneous catalyst into a heterogeneous one through the stabilization of active Pd sites onto the large surface area of inorganic support by an organic spacer to create organic–inorganic hybrid catalyst. A simple route is described to prepare a supported CN-palladacycle catalyst based on functionalized TiO2 as a very suitable inorganic support. The resulting ultra Pd-loaded material was investigated as a catalyst for copper- and amine-free Sonogashira coupling reactions of phenylacetylene with aryl halides which were carried out in the absence of phosphine ligands under air. In the context of green chemistry, heterogeneous catalysis of Sonogashira coupling reactions of phenylacetylene with aryl iodides carried out in ethanol as a green solvent.
2. Results and discussions
2.1. Synthesis and characterization of chloro-bridged CN-palladacycle
Nitrogen-derived palladacycles, have attracted much attention as exciting catalyst precursors for cross coupling reactions due to their accessibility, thermal stability, slow decomposition and high catalytic activity.11,43New dinuclear CN-palladacycle was synthesized through the reaction of palladium(II) acetate with bis[4-(dimethylamino)phenyl]methaniminium chloride (Auramine-O) in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio (Scheme 1). The green orthopalladated chloro-bridged complex could be produced directly from the refluxing reaction of Pd(OAc)2 with Auramine-O ligand in toluene for one 24 hours (method A). However, during the second method (B), the reaction of Pd(OAc)2 with Auramine-O ligand was carried out in THF, at room temperature for higher reaction time of 72 hours to afford the pure product in higher yield. The dinuclear CN-palladacycle was characterized by IR and NMR spectroscopies. The elemental analysis result of the prepared compound was in good agreement with the calculated values.
:1 molar ratio (Scheme 1). The green orthopalladated chloro-bridged complex could be produced directly from the refluxing reaction of Pd(OAc)2 with Auramine-O ligand in toluene for one 24 hours (method A). However, during the second method (B), the reaction of Pd(OAc)2 with Auramine-O ligand was carried out in THF, at room temperature for higher reaction time of 72 hours to afford the pure product in higher yield. The dinuclear CN-palladacycle was characterized by IR and NMR spectroscopies. The elemental analysis result of the prepared compound was in good agreement with the calculated values.
 | ||
| Scheme 1 Synthetic method for new orthometallated compound. (i) Toluene, Δ, 24 h; (ii) THf, 72 h. | ||
As illustrated in Fig. 1, the FT-IR spectrum of Auramine-O ligand shows the peak at 1685 cm−1 due to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretching vibration.44 This band appears at the lower energies (1606 cm−1) in the spectrum of synthesized dinuclear complex (Fig. 1b) indicating the coordination of the ligand to the metal. As clearly shown in Fig. 1a, the iminium salt displays strong, broad N–H stretching absorptions in the 2250 to 3000 cm−1 region, where overlap with C–H absorption occurs. The lower frequency region of FT-IR spectrum shows the characteristic bands at 674 and 416 cm−1 related to the ν(Pd–C) and ν(Pd–N) vibrations in the dinuclear complex, respectively (Fig. 1b).45,46 These vibrational bands confirm the successful coordination of the CN-orthopalladation of Auramine-O ligand to the palladium(II) center by covalent interaction.
N stretching vibration.44 This band appears at the lower energies (1606 cm−1) in the spectrum of synthesized dinuclear complex (Fig. 1b) indicating the coordination of the ligand to the metal. As clearly shown in Fig. 1a, the iminium salt displays strong, broad N–H stretching absorptions in the 2250 to 3000 cm−1 region, where overlap with C–H absorption occurs. The lower frequency region of FT-IR spectrum shows the characteristic bands at 674 and 416 cm−1 related to the ν(Pd–C) and ν(Pd–N) vibrations in the dinuclear complex, respectively (Fig. 1b).45,46 These vibrational bands confirm the successful coordination of the CN-orthopalladation of Auramine-O ligand to the palladium(II) center by covalent interaction.
 | ||
| Fig. 1 FT-IR spectra of: (a) Auramine-O ligand, (b) dinuclear palladium complex (ADP) (c) TiO2, (d) fresh TiO2-supported Pd catalyst. | ||
In the 1H NMR spectrum, the signal related to the methyl protons of cyclometallated Auramine-O appears at 3.00 ppm. All of the 1H NMR signals for the aromatic protons of the complex are shifted with respect to the parent iminium salt confirming the coordination of Auramine-O.
2.2. Synthesis and characterization of functionalized TiO2-supported Pd catalyst
Organic–inorganic hybrid catalysts simultaneously have the advantage of easy separation and recovery of the heterogeneous catalysts in addition to homogeneous reaction conditions. They are composed of three components including support, organic spacer and an active center.47,48In order to design heterogeneous catalysts, solid supports containing nitrogen as donor atoms have been achieved special interest. Since N-donor ligands are air stable, less expensive and nontoxic, they can be used to replace phosphine groups and coordinate to the various transition metal complexes.
Due to the fact that inorganic supports like TiO2 show advantages such as mechanical stability and resistance against solvent, chemical reagents and high temperature,49 a general rout has been designed to support a CN-palladacyclic complex onto the organically modified-TiO2 surface to prepare an efficient heterogeneous pre-catalyst. The synthetic procedure is illustrated in Scheme 2.
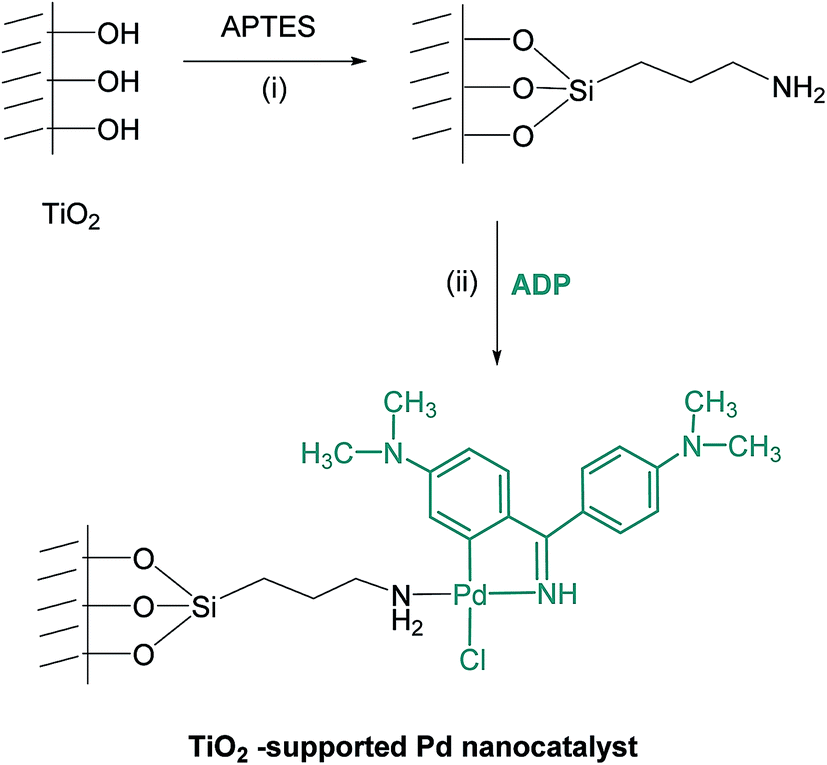 | ||
| Scheme 2 Synthetic method and proposed route for the synthesis of new TiO2-supported catalyst. (i) Dry toluene, 12 h, reflux (ii) acetone, 24 h. | ||
TiO2 usually shows pH-dependent surface charges in the aqueous solution because of the existence of Ti–OH on the surface. The absorption property of TiO2 greatly changes with environment of different pH value. Considering that the catalytic properties of supported catalysts depend on their preparation conditions, we tried to prepare the ligand functionalized TiO2 using the pH-dependent procedure.50,51 To obtain the TiO2-supported Pd catalyst, a mixture of chloro-bridged orthopalladated complex (ADP) and a functionalized TiO2 were refluxed in acetone for 24 hours.
The stabilization of CN-palladacycle onto the functionalized TiO2 is due to donation of an electron lone pairs on the nitrogen groups of 3-aminopropyltrimethoxysilane (APTES) into the unoccupied orbital of the palladium atoms in continuous of the bridge cleavage reaction of the dimeric compound ADP (Scheme 2).
Fig. 1 shows the FT-IR spectra of Auramine-O ligand, Auramine-O dimeric palladacycle (ADP), TiO2 and fresh TiO2-supported Pd catalyst. Corresponding to the FT-IR spectrum of TiO2-supported catalyst illustrated in Fig. 1d, the broad band centered at 500–600 cm−1 is likely due to the vibration of the Ti–O bonds in the TiO2 lattice.52 The peak at 1627 cm−1 and the broad peak appearing at 3451 cm−1 are assigned to vibrations of hydroxyl groups.45,52,53 In modified sample these peaks overlap with broad bands of the stretching and deformation modes of NH2 groups.45,54 The stretching vibrations of aliphatic groups are appeared at 2951 cm−1. The bands at 1124 and 1383 cm−1 are attributed to the characteristic absorptions of the Si–CH2 and Si–O groups, respectively.55 Also, the presence of several bands with medium intensity in the 2839–3062 cm−1 regions is allocated to C–H stretching of groups (symmetric and asymmetric stretching).
The elemental analysis further proved the existence of C, H, and N atoms in the prepared TiO2-supported Pd catalyst.
Powder X-ray diffraction (XRD) analysis for fresh catalyst exhibits the sharp reflections belonging to the TiO2 nanoparticles in the anatase phase (Fig. 2). Although no Pd species could be detected by XRD pattern due to the low amount of Pd content or its high dispersion on the solid surface, the presence of Pd species was clearly evidenced by X-ray photoelectron spectroscopy (XPS) verifying the orthopalladated complex ADP coated on the TiO2.
 | ||
| Fig. 2 Powder XRD pattern of TiO2-supported Pd catalyst. | ||
Fig. 3 shows high resolution XPS spectra of TiO2-supported Pd catalyst. The Pd 3d spectrum in Fig. 3a related to the fresh catalyst, shows the characteristic Pd 3d5/2 and 3d3/2 doublet peaks, with a peak separation of 5.26 eV. The fitted 3d5/2 peaks are located near the expected positions for the Pd0 and Pd2+ species at around 335.3 (ref. 56–58) and 337.6 eV,56,57 respectively. The fitting indicates that Pd in the fresh catalyst is a mixture of Pd2+ and Pd0 states. After the first run, Pd0 is observed in higher proportions, which indicates that after the catalytic cycle Pd2+ cations were transformed into Pd0 (Fig. 3b). According the Pd 3d spectrum of Pd catalyst after the forth catalytic cycle (Fig. 3c), presence of the characteristic Pd 3d5/2 and 3d3/2 doublet peaks related to the Pd0 species confirming the conversion of the whole Pd2+ ions to Pd0. In fact, the percentage of Pd2+ decreases from 53% for fresh catalyst to 36% from first run and approximately 0% for the fourth run as we can see in Fig. 3. According to the XPS data of Pd catalyst before and after the recycle, the Si 2p spectrum shows only one component, with the Si 2p peak position around 103.3 eV, assigned to Si–O bonds.58 The Ti 2p spectrum shows the characteristic Ti 2p3/2 and 2p1/2 doublet peaks, with a peak separation of 5.54 eV. The characteristic shake-up peaks for TiO2 are also observed. The fitted 2p3/2 peak is located near the expected positions for the TiO2 species at around 458.7 eV.58 The HR-XPS N 1s spectra of the fresh Pd catalyst shows two components, one at around 400.0 eV attributed to C–N bonds58 and another minor component at around 401.9 eV attributed to N+–H bonds.59 The N 1s fitted spectrum after the first recycle shows one component at around 400.0 eV attributed to C–N bonds58 and after the forth recycle, the nitrogen presented a very low signal and the peak was not possible to fit. The Cl 2p spectrum of the fresh Pd catalyst shows the characteristic Cl 2p3/2 and 2p1/2 doublet peaks, with a peak separation of 1.6 eV. After the recycle, no chlorine signal is detected which indicate that after the catalytic cycles Pd2+ species were transformed into Pd0 and the ligand was removed from palladacycle.
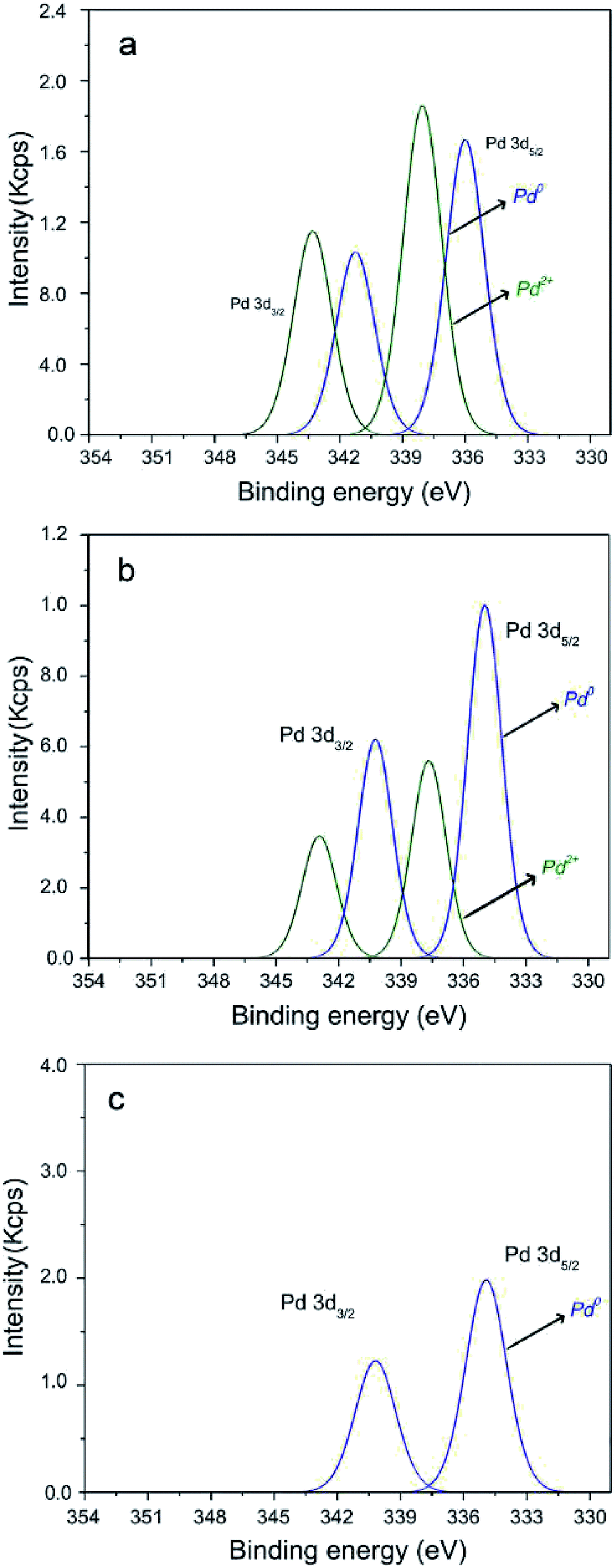 | ||
| Fig. 3 XPS spectra of high resolution Pd 3d of (a) fresh TiO2-supported Pd catalyst, (b) after the first run and (c) after the forth run. | ||
As illustrated in the similar investigations60 the formation of TiO2-supported Pd(0) nanoparticles are possibly due to the donation of an electron lone pairs on the nitrogen groups into an unoccupied orbital of the palladium nanoparticles. Moreover, the presence of ethanol as solvent, possibly leads to the reduction of Pd(II) to Pd(0).
XPS analysis showed that the palladium content of fresh catalyst was about 1.5%. The palladium content of catalyst after the reaction cycle was considerable, according to the XPS analysis. Further information about homogeneous or heterogeneous nature of the catalyst was obtained by hot filtration test which were in accordance with the results obtained from XPS analysis and confirmed that using this catalyst reaction proceeds mostly under heterogeneous conditions.
To obtain the morphology and particle size of the catalyst, FE-SEM images of the catalyst were carried out (Fig. 4). Furthermore, energy-dispersive X-ray spectroscopy (EDX) obtained from SEM shows the presence of Pd atoms as well as Ti, Si, N, and O atoms in the structure of fresh TiO2-supported Pd catalyst (Fig. 5).
 | ||
| Fig. 4 FE-SEM image of fresh TiO2-supported Pd catalyst. | ||
 | ||
| Fig. 5 EDX spectrum of the fresh TiO2-supported Pd catalyst. | ||
According to the TEM images, the Pd nanoparticles are formed and well dispersed through the supported inorganic surface. It was verified that synthesis of catalyst through the pH adsorption method played an important role in the distribution of nanoparticles. The size distribution of fresh TiO2-supported Pd catalyst is shown in Fig. 6. The histogram could be fitted to a Gaussian function, yielding a mean size of 2.24 nm based on the sizes of 90 particles (Fig. 6a). According to the TEM images after the first run, the average particle size of Pd0 in supported Pd catalyst was 2.61 nm. The average particle size of Pd0 after the forth run was 3.31 nm.
 | ||
| Fig. 6 TEM images of (a) fresh TiO2-supported Pd catalyst, (b) after the first run and (c) after the forth run. The histograms illustrate the size distribution of Pd catalyst. | ||
2.3. Sonogashira reactions catalyzed by Pd NPs
There are several disadvantages in the earlier reported methods for the Sonogashira coupling reactions such as employing expensive, toxic and air/moisture sensitive phosphine ligands, long reaction times, low yields, inert conditions and environmental pollution caused by the formation of side products and use of large amount of amines as organic solvents or bases.61 In the copper-mediated Sonogashira coupling reactions, the Glaser-type oxidative dimerization of the terminal alkynes cannot be avoided62 which produce undesired side products (diynes) that are generally difficult to separate. Consequently, in our screening experiments, the realization of a Sonogashira reaction catalyzed by the low Pd-loading of TiO2-supported Pd catalyst in the absence of copper, amine and phosphine ligands was our goal.The effectiveness of supported Pd catalyst for the Sonogashira cross-coupling reaction of iodobenzene with phenylacetylene was initially investigated. First we investigated the effect of solvent in the Sonogashira reaction of iodobenzene with phenylacetylene (Table 1, entry 1–7). The yield was significantly decreased when H2O was used as the solvent (Table 1, entry 5); this may be due to the insolubility of the substrate and catalyst in water. However, application of 1:1 aqueous EtOH improves the yield to some extent (Table 1, entry 6). On the other hand inferior results were obtained with solvents like THF, CH3CN, toluene and DMF (Table 1, entries 1–4). As shown in Table 1 better performance of the catalytic system was observed when EtOH was used and based on the above we have considered EtOH for further optimization. A very low Pd-loading of 0.005 mol% was found to be the optimum amount of catalyst. To investigate the effect of the base which plays a crucial role in the overall performance of a catalytic system in a Sonogashira reaction, several inorganic and organic bases were tested (Table 1, entry 7–10). K2CO3 was found to be reactive in our catalytic system and we have considered it for further optimization. The preliminary investigation for reaction optimization was performed with 0.005 mol% of Pd catalyst and two equivalent of K2CO3 in EtOH (4 ml) at 85 °C under aerobic condition. The results obtained are highlighted in Table 1. The turnover frequency of the catalyst was defined to be 5880, which was regarded as a factor to show the high efficiency of the catalyst.
|
||||||
|---|---|---|---|---|---|---|
| Entry | [Pd] (mol%) | Solvent | Base | Time | Temperature (°C) | Conversiona (%) |
| a Reactions were monitored using GC. | ||||||
| 1 | 0.005 | DMF | K2CO3 | 6 h | 85 | 60 |
| 2 | 0.005 | CH3CN | K2CO3 | 6 h | 85 | 41 |
| 3 | 0.005 | Toluene | K2CO3 | 4 h | 85 | Trace |
| 4 | 0.005 | THF | K2CO3 | 4 h | 85 | Trace |
| 5 | 0.005 | H2O | K2CO3 | 4 h | 85 | Trace |
| 6 | 0.005 | Ethanol:H2O (1:1) |
K2CO3 | 4 h | 85 | 70 |
| 7 | 0.005 | Ethanol | K2CO3 | 30 min | 85 | 99 |
| 8 | 0.005 | Ethanol | Na2CO3 | 6 h | 85 | 43 |
| 9 | 0.005 | Ethanol | K3PO4·3H2O | 6 h | 85 | 90 |
| 10 | 0.005 | Ethanol | NaOAc | 4 h | 85 | 10 |
| 11 | 0.005 | Ethanol | K2CO3 | 6 h | R. T. | Trace |
| 12 | 0.02 | Ethanol | K2CO3 | 15 min | 85 | 99 |
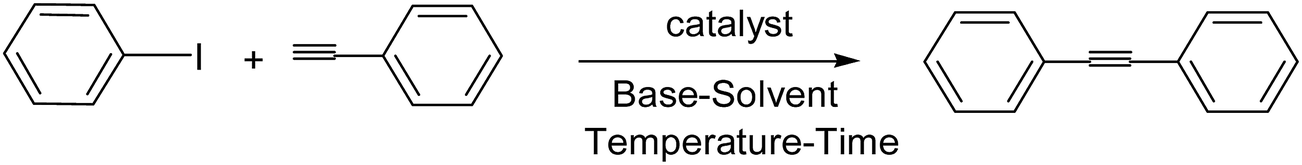
To demonstrate the versatility of the catalytic system, we investigated the reaction using a variety of aryl iodide with phenylacetylene under the optimized conditions. It was observed that aryl iodides underwent cross coupling within a short reaction time at very low catalyst Pd-loading of 0.005 mol%, with excellent yields (Table 2). They may produce very high TOFs in such catalytic reactions.
|
|||||
|---|---|---|---|---|---|
| Entry | R | Time | Conversion (%) | TONb | TOFc (h−1) |
| a Reaction conditions: aryl halides (1.0 mmol), phenylacetylene (1.2 mmol), K2CO3 (2.0 mmol), EtOH (4 ml), catalyst (0.005 mol% [Pd]), 85 °C. Reactions were monitored using GC.b TON = mmol of products/mmol of catalyst.c TOF = TON/time. | |||||
| 1 | H | 30 min | 98 | 5880 | 5880 |
| 2 | p-NO2 | 20 min | 99 | 5940 | 11880 |
| 3 | m-NO2 | 1 h | 90 | 5400 | 2700 |
| 4 | p-MeO | 2 h | 99 | 5940 | 5940 |
| 5 | m-MeO | 3 h | 99 | 5940 | 1980 |

To study the performance of TiO2-supported Pd catalyst for the Sonogashira cross-coupling reaction of aryl bromide with phenylacetylene, optimization of the reaction conditions with various solvents and catalyst dosages were applied for bromobenzaldehyde in different range of time reactions and temperatures (Table 3). Among the different reaction conditions, maximum conversion was observed when we have considered the reaction in DMF (4 ml), in the presence of 0.02 mol% of Pd catalyst at 110 °C, under aerobic condition for 8 hours.
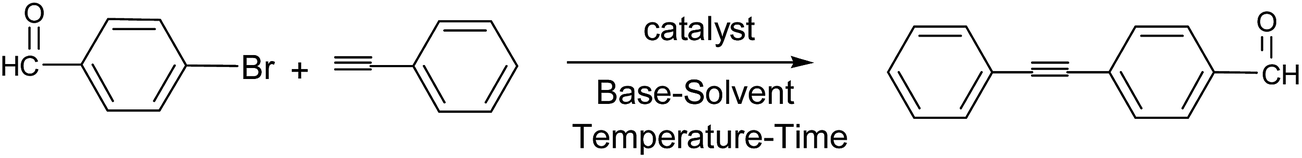
The generality of the current system has been investigated with several electronically diverse aryl bromide and phenylacetylene (Table 4, entries 1–8). Different functionality at the phenyl ring of the aryl halide moiety affects the reaction conditions. When the reactions of activated aryl bromides containing electron deficient phenyl rings with phenylacetylene were carried out in DMF at 110 °C for 8 h, excellent yields of desired cross-coupling products were observed. However, the electron donating groups at the para position of aryl bromides couple smoothly with phenylacetylene to give good to excellent yield of cross coupling products. Considering the steric effect, aryl bromides with ortho substituents, provided the corresponding products in high yields (Table 4, entry 4).
|
|||||
|---|---|---|---|---|---|
| Entry | R | X | Conversionb (%) | TON | TOFc (h−1) |
| a Reaction conditions: aryl halides (1.0 mmol), phenylacetylene (1.2 mmol), K2CO3 (2.0 mmol), DMF (4 ml), catalyst (0.02 mol% [Pd]), 110 °C. Reactions were monitored using GC and TLC.b TON = mmol of products/mmol of catalyst.c TOF = TON/time. | |||||
| 1 | H | Br | 50 | 750 | 93.75 |
| 2 | p-CHO | Br | 95 | 1425 | 285 |
| 3 | p-MeCO | Br | 94 | 1410 | 235 |
| 4 | o-NO2 | Br | 97 | 1455 | 181.875 |
| 5 | m-Me | Br | 10 | 150 | 18.75 |
| 6 | 1-Bromonaphthalene | 30 | 450 | 56.25 | |
| 7 | 2-Bromopyridine | 50 | 750 | 93.75 | |
| 8 | 2-Bromothiophene | 57 | 855 | 106.875 | |
| 9 | H | Cl | 50 | 750 | 93.75 |
| 10 | p-NO2 | Cl | 62 | 930 | 116.25 |
| 11 | p-NH2 | Cl | 28 | 420 | 52.5 |
| 12 | o-OH | Cl | 34 | 510 | 63.75 |

Finally Sonogashira reactions of phenylacetylene and aryl chlorides were also investigated in our present system, under the optimized condition used for the case of aryl bromides. To check the effectiveness of our protocol, phenylacetylene was reacted with electron rich 4-aminobenzene and electron deficient nitrochlorobenzene. Although aryl chlorides are not so reactive and are employed less in palladium-catalyzed coupling reactions,63–65 modest to good yield of desired cross coupling products was observed in all cases (Table 4, entries 9–12).
A possible catalytic reaction cycle for the Sonogashira reaction has been shown in Scheme 3, over the active Pd-sites present in the TiO2-supported heterogeneous catalyst. At first, oxidative addition of aryl halide to the Pd catalyst takes place, followed by transmetallation of alkyne and lastly, reductive elimination of the final product.
 | ||
| Scheme 3 Possible catalytic cycle of the heterogeneously catalyzed Cu-free Sonogashira reaction over TiO2-supported Pd catalyst. | ||
Aryl halides containing electron-withdrawing groups improve the oxidative addition of C–X to the Pd catalyst, as an activated halo derivatives.25 However, electron-rich aryl halides require harsher experimental conditions to give acceptable results. Although, the mechanism of the Sonogashira reactions has not yet been established clearly, we assume that the lower tendency of electron-rich aryl halides towards metallo-oxidative insertions could be responsible for this diminished reactivity.66,67
Although, there is no second metal in the copper-free Sonogashira reaction, the second step is conventionally called transmetallation. Several studies have attempted to understand how the alkyne is transferred to the Pd catalyst through the transmetallation step. Amines generally used in the Sonogashira reaction, appreciably deprotonated the alkyne in solution. But despite the fact that the Sonogashira reaction is occurred in the amine-free condition in this study, we cannot have the deprotonation of the alkyne in solution. Therefore, deprotonation is hypothesized to occur on the Pd center. Initially, the alkyne coordinates to the Pd center in the η2-fashion, pulling electron density away from the acetylene. This makes the terminal proton more acidic, enabling deprotonation by weaker bases (Scheme 3).68–70 Importantly, the electronic properties of the terminal alkyne have considerable effects the on the deprotonation mechanism: electron withdrawing groups (EWG) decrease and electron donating groups (EDG) increase the energy required for deprotonation.68 The base initially forms the more reactive species of the type Pd0L2 in the oxidative addition step lead to accelerate the overall reaction.70 Using of stronger bases, an excess amount of phenylacetylene and more acidic phenylacetylenes considerably increase the rate by affecting the deprotonation step.71
Furthermore, the reaction kinetics of Sonogashira reaction of phenylacetylene with iodobenzene with 0.02 mol% catalyst was investigated and the result is shown in Fig. 7. A control experiment indicated that the coupling reaction did not occur smoothly in the absence of a catalyst. The yield of the product increased quickly with reaction time until it reached 96% at the 30 minutes.
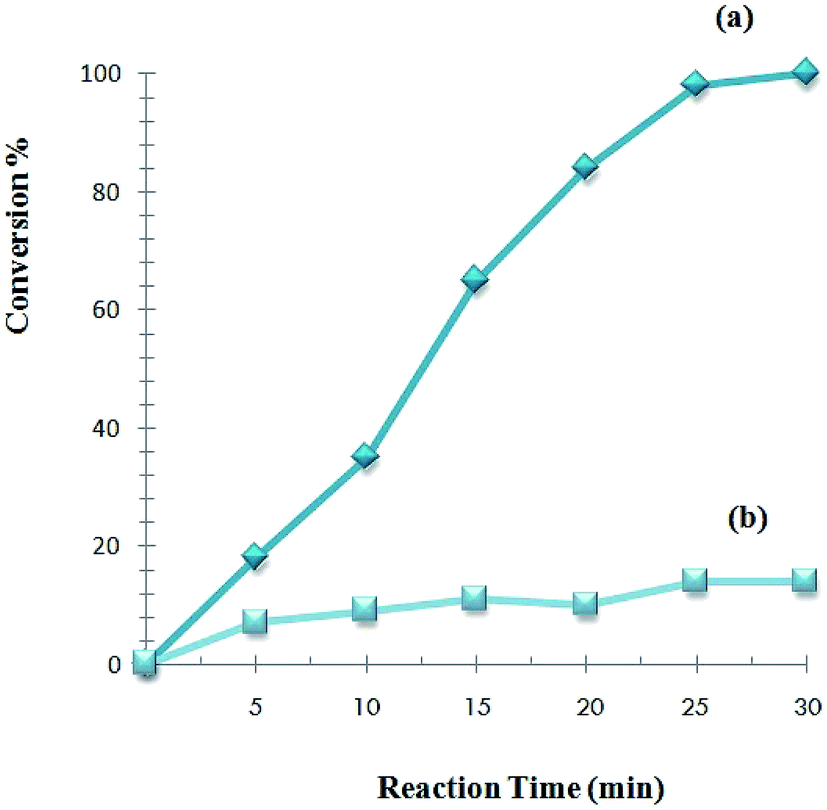 | ||
| Fig. 7 Kinetic profiles of the Pd catalyst in the Sonogashira reaction. (a) Normal reaction kinetics in the presence of catalyst; (b) reaction kinetics in the absence of catalyst. Reaction conditions: iodobenzene (1.0 mmol), phenylacetylene (1.2 mmol), K2CO3 (2.0 mmol), EtOH (4 ml), catalyst (0.02 mol% Pd), 85 °C, 30 min. | ||
2.4. Comparison with the other studies
To further evaluate the catalytic performance of catalyst, a comparison was made with several literature-reported supported catalysts (Table 5). The reactions of iodobenzene with phenylacetylene were taken as the standard reactions and the results are listed in Table 5. It can be clearly seen that for the Sonogashira reaction of iodobenzene with phenylacetylene, the current catalyst is superior to others in terms of catalytic efficiency. Moreover, this catalyst also proved to be the most efficacious in activity for the Sonogashira reaction, as reflected from its high TOF value. It should be noted that the supported-Pd catalyzed Sonogashira reactions of iodobenzene with phenylacetylene were carried out in EtOH in the absence of organic solvents, which makes this catalytic capacity more impressive. Thus this catalytic system provides a simple and green method for the Sonogashira reaction of aryl iodides. Compared to previous research,72–77 this catalytic system requires a very small amount of catalyst Pd-loading and can be run under relatively mild reaction conditions.
|
|||||
|---|---|---|---|---|---|
| Entry | Catalysta | Temp. (°C)/time (h) | Medium | Yield (%) | Ref. |
| a Data in parentheses indicate mol% of Pd used. | |||||
| 1 | MMT@Pd/Cu (1) | 65/24 | DME/H2O | 97 | 72 |
| 2 | Pd@Fe3O4 (0.5) | 110/24 | DMF | 84 | 73 |
| 3 | PNP–SSS (1.2) | 100/3 | H2O | 95 | 74 |
| 4 | FDU–NHC/Pd(II) (1) | 100/3 | DMA | 94 | 75 |
| 5 | CS/MMT/Pd (0.3) | 110/5 | DMSO | 94 | 76 |
| 6 | Si–P4VPy–Pd (0.5) | 120/0.25 | NMP | 90 | 77 |
| 7 | Pd/APTES/TiO2 (0.005) | 85/0.5 | EtOH | 99 | This study |
2.5. Recyclability
The reusability of the catalysts is a very important theme and makes them useful for commercial applications. After the catalytic reactions, the recovery and reusability of TiO2-supported Pd catalyst were initially investigated in the Sonogashira reaction of iodobenzene with phenylacetylene under the same condition. Before running another cycle, the solid was separated by simple filtration and washed with acetone, water and then dried. The catalyst is reusable and used for four runs. The results are shown in Fig. 8. | ||
| Fig. 8 A recyclability test for catalyst Sonogashira cross-coupling reaction of iodobenzene with phenylacetylene (reaction condition are the same as given in Table 2). | ||
3. Experimental
3.1. General
1H NMR spectra were recorded at room temperature on a 400 MHz Bruker spectrometer. Chemical shifts (δ) are reported relative to internal TMS. C, H and N elemental analysis were performed using a PE 2400 series analyzer. Fourier transform infrared (FT-IR) spectra were obtained in KBr pellets with a Jasco FT/IR 680 plus instrument. Scanning electron microscopy (SEM) studies were carried out at 15 kV using a HITACHI S-4160 instrument (Japan). Energy dispersive X-ray analysis (EDX) results were obtained at 20 kV using Philips (Model XL-30) instrument. Transmission electron micrographs (TEM) were obtained with a JEOL JEM 1010 microscope operating at an accelerating voltage of 100 kV. The X-ray powder diffraction (XRD) pattern was recorded using a Scintag X-ray diffractometer with a 1.54 Å (Cu Kα) X-ray radiation source. The XPS experiments were performed in a SPECS Sage HR 100 spectrometer with a non-monochromatic X-ray source (magnesium Kα line of 1253.6 eV energy and a power applied of 250 W), placed perpendicular to the analyzer axis and calibrated using the 3d5/2 line of Ag with a full width at half maximum (FWHM) of 1.1 eV. An electron flood gun was used during measurements to neutralize charging effects. The selected resolution for the spectra of the different elements was 15 eV of pass energy and 0.15 eV per step. All measurements were made in an ultra high vacuum (UHV) chamber at pressure around 8 × 10−8 mbar. In the fittings Gaussian–Lorentzian functions were used (after a Shirley background correction). Analytical TLC was performed using Merck TLC silica gel 60 F254 glass plates. Gas chromatographic (GC) analyses were performed using an Agilent Technologies 6890N chromatograph equipped with a flame ionization detector (FID) and an HB 50+ column (length = 30 m, inner diameter 320 μm, and film thickness = 0.25 μm). The temperature program for the GC analysis was from 70 to 200 °C at 20 °C min−1, held at 200 °C for 0 min, heated from 200 to 280 °C at 10 °C min−1 and held at 280 °C for 1 min. The inlet and detector temperatures were set at 260 °C and 280 °C respectively. Products were identified by comparison with authentic samples. All the reactants and solvents were used as commercially available chemicals without any purification.3.2. Preparation of Auramine-O dimeric palladacycle (ADP)
:5 CH2Cl2/n-hexane and cold Et2O to give dark-green chloro-bridge dimeric complex.:5 CH2Cl2/n-hexane and cold Et2O to give dark-green chloro-bridge dimeric complex.Yield (62–76%); anal. calc. for C34H24N6 Cl2Pd2: C, 51.03; H, 3.02; N, 10.50; found: C, 51.87; H, 2.98; N, 10.28; IR (KBr, cm−1): ν(Pd–N) = 416, ν(Pd–C) = 674, ν(CN) = 1606; 1H NMR (DMSO-d6, ppm): δ = 3.00 (d, 24H, aliphatic), 6.38 (d, 2H, aromatic), 6.81 (d, 4H, aromatic), 7.01 (d, 2H, aromatic), 7.41 (d, 4H, aromatic), 7.47 (s, 1H, aromatic), 8.94 (s, 1H, aromatic).
3.3. Preparation and characterization of the TiO2-supported Pd catalyst
TiO2 was activated by refluxing in concentrated hydrochloric acid (6 M) for 24 h and then washed thoroughly with the deionized water and dried before undergoing chemical surface modification. Refluxing the activated TiO2 (10 g) with 1.5 mmol 3-aminopropyltrimethoxysilane in dry toluene for 18 h. The solid materials were filtered off and washed with hot toluene and then dried in oven at 110 °C overnight to give the surface bound amine group. The catalyst was prepared by stirring a mixture of surface bound ligand L@TiO2 (4 g) and ADP (0.52 mmol, 0.117 g) in dry acetone (100 ml) at room temperature for 24 h. After stirring, the resulting greenish solid was filtered, washed with large volume of acetone, ethanol and ether. It was then dried in an oven at 95 °C overnight to furnish the corresponding catalyst.3.4. General experimental procedure for Sonogashira reaction with aryl iodides
A round bottomed flask equipped with a condenser was charged with aryl iodides (1.0 mmol), phenylacetylene (1.2 mmol), K2CO3 (2.0 mmol) and Pd catalyst (0.005 mol% of Pd) in EtOH (4 ml). Typical procedure for Sonogashira cross-coupling reaction was placed in an oil bath and the mixture was stirred and heated at 85 °C for 30 minutes.3.5. General experimental procedure for Sonogashira reaction with aryl bromides and chlorides
A round bottomed flask equipped with a condenser was charged with aryl bromides or chlorides (1.0 mmol), phenylacetylene (1.2 mmol), K2CO3 (2.0 mmol) and Pd catalyst (0.02 mol% of Pd) in DMF (4 ml). Typical procedure for Sonogashira cross-coupling reaction was placed in an oil bath and the mixture was stirred and heated at 110 °C for 8 h.3.6. General procedure for recycling reactions
When the corresponding Sonogashira reaction according to the procedure described in previous sections was finished, the catalyst was washed with acetone and water. It was dried under air and reused without any pretreatment for repeating cycles.3.7. Characterization of the products of Sonogashira cross-coupling reactions
4. Conclusion
A general approach has been designed to synthesize a new dinuclear chloro-bridged CN-palladacycle derived from bis[4-(dimethylamino)phenyl]methaniminium chloride (Auramine-O). In the next step, we have successfully developed a new heterogeneous TiO2-supported palladium catalyst originated from the CN-palladacycle precursor which is fully characterized with FT-IR, XRD, SEM, EDX, TEM and XPS techniques. This hybrid catalyst demonstrated high catalytic activities in the copper-, amine and phosphine-free Sonogashira coupling reactions of phenylacetylene with aryl halides (Cl, Br, I) in the presence of very low catalyst Pd-loading. This class of heterogeneous Pd catalyst allows the reaction of phenylacetylene and aryl iodides to promote with remarkable yields under environmentally green condition.Acknowledgements
This work was financially supported by the Isfahan University of Technology. We gratefully acknowledge the funding support received for this project from the Iranian Nano Technology Initiative Council.References
- M. López-Torres, A. Fernández, J. J. Fernández, A. Suárez, S. Castro-Juiz, J. M. Vila and M. Teresa Pereira, Organometallics, 2001, 20, 1350–1353 CrossRef.
- S.-W. Lai, T.-C. Cheung, M. C. W. Chan, K.-K. Cheung, S.-M. Peng and C.-M. Che, Inorg. Chem., 2000, 39, 255–262 CrossRef CAS PubMed.
- S.-H. Li, C.-W. Yu and J.-G. Xu, Chem. Commun., 2005, 450–452 RSC.
- V. Sicilia, J. Fornies, S. Fuertes and A. Martín, Inorg. Chem., 2012, 51, 10581–10589 CrossRef CAS PubMed.
- K. Karami, Z. Mehri Lighvan, S. Askari Barzani, A. Yeganeh Faal, M. Poshteh-Shirani, T. Khayamian, V. Eigner and M. Dusek, New J. Chem., 2015, 39, 8708–8719 RSC.
- K. Karami, M. Hosseini-Kharat, H. Sadeghi-Aliabadi, J. Lipkowski and M. Mirian, Eur. J. Med. Chem., 2014, 73, 8–17 CrossRef CAS PubMed.
- P.-K. Chow, W.-P. To, K.-H. Low and C.-M. Che, Chem.–Asian J., 2014, 9, 534–545 CrossRef CAS PubMed.
- M. V. Kulikova, K. P. Balashev, P.-I. Kvam and J. Songstad, Russ. J. Gen. Chem., 2000, 70, 163–170 CAS.
- C.-W. Yu, S.-H. Li, H. Zheng and J.-G. Xu, Chin. J. Chem., 2007, 25, 797–801 CrossRef CAS.
- D. Aguilar, M. A. Aragues, R. Bielsa, E. Serrano, T. Soler, R. Navarro and E. P. Urriolabeitia, J. Organomet. Chem., 2008, 693, 417–424 CrossRef CAS.
- D. A. Alonso and C. Nájera, Chem. Soc. Rev., 2010, 39, 2891–2902 RSC.
- I. P. Beletskaya and A. V. Cheprakov, J. Organomet. Chem., 2004, 689, 4055–4082 CrossRef CAS.
- M. Mondal and U. Bora, New J. Chem., 2016, 40, 3119–3123 RSC.
- M. Micksch, M. Tenne and T. Strassner, Organometallics, 2014, 33, 3966–3976 CrossRef CAS.
- N. Marion, O. Navarro, J. G. Mei, E. D. Stevens, N. M. Scott and S. P. Nolan, J. Am. Chem. Soc., 2006, 128, 4101 CrossRef CAS PubMed.
- S. O. Kang, M. A. Hossain and K. Bowman-James, Coord. Chem. Rev., 2006, 250, 3038–3052 CrossRef CAS.
- S. O. Kang, R. A. Begum and K. Bowman-James, Angew. Chem., Int. Ed., 2006, 118, 8048–8061 CrossRef.
- E. Negishi and L. Anastasia, Chem. Rev., 2003, 103, 1979–2017 CrossRef CAS PubMed.
- K. C. Nicolaou, P. G. Bulger and D. Sarlah, Angew. Chem., Int. Ed., 2005, 44, 4442–4489 CrossRef CAS PubMed.
- A. de Meijere and F. Diederich, Metal-Catalyzed Cross-Coupling Reactions, Wiley-VCH, Weinheim, 2004 Search PubMed.
- L. Yin and J. Liebscher, Chem. Rev., 2007, 107, 133–173 CrossRef CAS PubMed.
- H. Doucet and J.-C. Hierso, Angew. Chem., Int. Ed., 2007, 46, 834–871 CrossRef CAS PubMed.
- C. Nájera and R. Chinchilla, Chem. Rev., 2007, 107, 874–922 CrossRef PubMed.
- K. Sonogashira, B. M. Trost and I. Fleming, Comprehensive Organic Synthesis, Pergamon Press, New York, 1991, vol. 3, p. 521 Search PubMed.
- K. Sonogashira, E. Negishi and A. de Meijere, Handbook of Organopalladium Chemistry for Organic Synthesis, Wiley-Interscience, New York, 2002 Search PubMed.
- P. Siemsen, R. C. Livingston and F. Diederich, Angew. Chem., Int. Ed., 2000, 39, 2632–2657 (Angew. Chem., 2000, 112, 2740–2767) CrossRef CAS.
- J. H. Li, Y. Liang and X. D. Zhang, Tetrahedron, 2005, 61, 1903–1907 CrossRef CAS.
- S. Thorand and N. Krause, J. Org. Chem., 1998, 63, 8551–8553 CrossRef CAS.
- A. Elangovan, Y. H. Wang and T. I. Ho, Org. Lett., 2003, 5, 1841–1844 CrossRef CAS PubMed.
- (a) K. Köhler, R. G. Heidenreich, J. G. E. Krauter and J. Pietsch, Chem.–Eur. J., 2002, 8, 622–631 CrossRef; (b) G. V. Ambulgekar, B. M. Bhanage and S. D. Samant, Tetrahedron Lett., 2005, 46, 2483–2485 CrossRef CAS.
- (a) C. M. Crudden, M. Sateesh and R. Lewis, J. Am. Chem. Soc., 2005, 127, 10045–10050 CrossRef CAS PubMed; (b) B. W. Glasspoole, J. D. Webb and C. M. Crudden, J. Catal., 2009, 265, 148–154 CrossRef CAS.
- (a) S. S. Pröckl, W. Kleist, M. A. Gruber and K. Köhler, Angew. Chem., Int. Ed., 2004, 43, 1881–1882 CrossRef PubMed; (b) M. J. Climent, A. Corma, S. Iborra and M. Mifsud, Adv. Synth. Catal., 2007, 349, 1949–1954 CrossRef CAS.
- (a) L. Djakovitch and K. Köhler, J. Am. Chem. Soc., 2001, 123, 5990–5999 CrossRef CAS PubMed; (b) M. Choi, D.-H. Lee, K. Na, B.-W. Yu and R. Ryoo, Angew. Chem., Int. Ed., 2009, 48, 3673–3676 CrossRef CAS PubMed.
- Z. Yinghuai, S. Chee Peng, A. Emi, S. Zhenshun, Monalisa and R. A. Kemp, Adv. Synth. Catal., 2007, 349, 1917–1922 CrossRef.
- B. M. Choudary, S. Madhi, N. S. Chowdari, M. L. Kantam and B. Sreedhar, J. Am. Chem. Soc., 2002, 124, 14127–14136 CrossRef CAS PubMed.
- K. Mori, K. Yamaguchi, T. Hara, T. Mizugaki, K. Ebitani and K. Kaneda, J. Am. Chem. Soc., 2002, 124, 11572–11573 CrossRef CAS PubMed.
- (a) Á. Molnár, A. Papp, K. Miklós and P. Forgo, Chem. Commun., 2003, 2626–2627 RSC; (b) S. Niembro, A. Shafir, A. Vallribera and R. Alibés, Org. Lett., 2008, 10, 3215–3218 CrossRef CAS PubMed.
- (a) K.-M. Choi, T. Akita, T. Mizugaki, K. Ebitani and K. Kaneda, New J. Chem., 2003, 27, 324–328 RSC; (b) K. Ebitani, Y. Fujie and K. Kaneda, Langmuir, 1999, 15, 3557–3562 CrossRef CAS; (c) T. Nishimura, N. Kakiuchi, M. Inoue and S. Uemura, Chem. Commun., 2000, 1245–1246 RSC.
- K. Karami, N. Haghighat-Naeini, V. Eigner, M. Dusek, J. Lipkowski, P. Hervés and H. Tavakol, RSC Adv., 2015, 5, 102424–102435 RSC.
- K. Karami, M. Ghasemi and N. Haghighat-Naeini, Catal. Commun., 2013, 38, 10–15 CrossRef CAS.
- K. Karami, Z. Karami-Moghadam and M. Hosseini-Kharat, Catal. Commun., 2014, 43, 25–28 CrossRef CAS.
- K. Karami and N. Haghighat Naeini, Appl. Organomet. Chem., 2015, 29, 33–39 CrossRef CAS.
- Palladacycles: Synthesis, Characterization and Applications, ed. J. Dupont and M. Pfeffer, Wiley-VCH, Weinheim, 2008 Search PubMed.
- L. Marin, B. Simionescu and M. Barboiu, Chem. Commun., 2012, 48, 8778–8780 RSC.
- K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds, John Wiley & Sons, New York, 1986 Search PubMed.
- N. N. Greenwood, Spectroscopic Properties of Inorganic and Organometallic Compounds, Royal Society of Chemistry, vol. 5, 1972 Search PubMed.
- P. Panster and S. Wieland, Applied Homogeneous Catalysis with Organometallic Compounds, ed. B. Cornils and W. A. Hermann, VCH, Weinheim, 1996, vol. 2, pp. 605–623 Search PubMed.
- B. Karimi, A. Zamani, S. Abedia and J. H. Clark, Green Chem., 2009, 11, 109–119 RSC.
- R. Daghrir, P. Drogui and D. Robert, Ind. Eng. Chem. Res., 2013, 52, 3581–3599 CAS.
- Z. Li, J. Chen, W. Su and M. Hong, J. Mol. Catal. A: Chem., 2010, 328, 93–98 CrossRef CAS.
- K. Karami, M. Bahrami Shehni and N. Rahimi, Appl. Organomet. Chem., 2013, 27, 437–443 CrossRef CAS.
- Y. Gao, Y. Masuda, Z. Peng, T. Yonezawa and K. Koumoto, J. Mater. Chem., 2003, 13, 608–613 RSC.
- B. Klingenberg and M. A. Vannice, Chem. Mater., 1996, 8, 2755–2768 CrossRef CAS.
- V. N. Khabashesku, J. L. Zimmerman and J. L. Margrave, Chem. Mater., 2000, 12, 3264–3270 CrossRef CAS.
- R. Tian, O. Seitz, M. Li, W. Hu, Y. J. Chabal and J. Gao, Langmuir, 2010, 26, 4563–4566 CrossRef CAS PubMed.
- G. Ding, W. Wang, T. Jiang and B. Han, Green Chem., 2013, 15, 3396–3403 RSC.
- J. Yang, D. Wang, W. Liu, X. Zhang, F. Bian and W. Yu, Green Chem., 2013, 15, 3429–3437 RSC.
- J. F. Moulder, W. F. Stickle, P. E. Sobol and K. D. Bomben, Handbook of X-ray Photoelectron Spectroscopy, Physical Electronics, 1995 Search PubMed.
- http://www.xpsfitting.com.
- Y. Borodko, S. M. Humphrey, T. D. Tilley, H. Frei and G. A. Somorjai, J. Phys. Chem. C, 2007, 111, 6288–6295 CAS.
- K. Sonogashira, in Comprehensive Organic Synthesis, ed. B. M. Trost and I. Fleming, Pergamon, New York, 1991, vol. 3 Search PubMed.
- S. Shylesh, V. Schunemann and W. R. Thiel, Angew. Chem., Int. Ed., 2010, 49, 3428–3459 CrossRef CAS PubMed.
- K. Selvakumar, A. Zapf and M. Beller, Org. Lett., 2002, 4, 3031–3032 CrossRef CAS PubMed.
- S. Prçckl, W. Kleist, M. A. Gruber and K. Kohler, Angew. Chem., Int. Ed., 2004, 43, 1881–1882 (Angew. Chem., 2004, 116, 1917–1918) CrossRef PubMed.
- K. Kçhler, W. Kleist and S. S. Prçckl, Inorg. Chem., 2007, 46, 1876–1883 CrossRef PubMed.
- C. Amatore, S. Bensalem, S. Ghalem, A. Jutand and Y. Medjour, Eur. J. Org. Chem., 2004, 366–371 CrossRef CAS.
- M. Bandini, R. Luque, V. Budarin and D. J. Macquarrie, Tetrahedron, 2005, 61, 9860–9868 CrossRef CAS.
- T. Ljungdahl, T. Bennur, A. Dallas, H. Emtenas and J. Martensson, Organometallics, 2008, 27, 2490–2498 CrossRef CAS.
- A. Soheili, J. Albaneze-Walker, J. A. Murry, P. G. Dormer and D. L. Hughes, Org. Lett., 2003, 5, 4191–4194 CrossRef CAS PubMed.
- S. Fujimori, T. F. Knopfel, P. Zarotti, T. Ichikawa, D. Boyall and E. M. Carreira, Bull. Chem. Soc. Jpn., 2007, 80, 1635–1657 CrossRef CAS.
- A. Tougerti, S. Negri and A. Jutand, Chem.–Eur. J., 2007, 13, 666–676 CrossRef CAS PubMed.
- W. Xu, H. Sun, B. Yu, G. Zhang, W. Zhang and Z. Gao, ACS Appl. Mater. Interfaces, 2014, 6, 20261–20268 CAS.
- M. Nasrollahzadeh, M. Maham, A. Ehsania and M. Khalaj, RSC Adv., 2014, 4, 19731–19736 RSC.
- A. Khalafi-Nezhad and F. Panahi, Green Chem., 2011, 13, 2408–2415 RSC.
- Y. Tao, L. Ying, Y. Chengfu, W. Haihong, L. Yueming and W. Peng, Chin. J. Catal., 2011, 32, 1712–1718 CrossRef.
- M. Zeng, X. Yuan, S. Zuo and C. Qi, RSC Adv., 2015, 5, 37995–38000 RSC.
- F. Farjadianan and B. Tamami, ChemPlusChem, 2014, 79, 1767–1773 Search PubMed.
- K. Karami and N. Haghighat Naeini, Turk. J. Chem., 2015, 39, 1199–1207 CrossRef CAS.
- A. R. Hajipour, E. Boostani and F. Mohammadsaleh, RSC Adv., 2015, 5, 94369–94374 RSC.
| This journal is © The Royal Society of Chemistry 2016 |