
Design, synthesis and effect of the introduction of a propargyloxy group on the fungicidal activities of 1-substituted phenoxypropan-2-amino valinamide carbamate derivatives†
Abstract
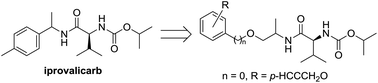
The cell walls of oomycetes are composed of cellulose, making cellulose synthase enzymes good targets for carboxylic acid amide fungicides. Valinamide carbamates are amino acid fungicides that represent excellent alternatives to conventional synthetic pesticides in terms of their ability to reduce the negative impacts of these compounds on human health and the environment. In a continuation of our research towards the development of new cellulose synthase inhibitors, we have developed a series of “stretched” analogues of iprovalicarb by the introduction of an additional OCH2 linker. The bioassay results indicated that compounds containing a small group at the para-position of phenyl gave excellent fungicidal activities with EC50 values ranging from 0.59 to 2.06 μmol L−1. Most notably, the introduction of a propargyloxy group led to a pronounced increase in the fungicidal activity. Furthermore, compound 7o bearing a propargyloxy group was identified as the most promising candidate because of its excellent fungicidal potency against oomycete diseases and good fungicidal activity against non-oomycete diseases.
 Please wait while we load your content...
Please wait while we load your content...

