DOI:
10.1039/C6RA17900B
(Paper)
RSC Adv., 2016,
6, 77887-77897
Isolation of lingzhifuran A and lingzhilactones D–F from Ganoderma lucidum as specific Smad3 phosphorylation inhibitors and total synthesis of lingzhifuran A†
Received
13th July 2016
, Accepted 9th August 2016
First published on 11th August 2016
Abstract
Lingzhifuran A (1) and lingzhilactones D–F (2–4), four new phenolic meroterpenoids were isolated from the fruiting bodies of Ganoderma lucidum. Their structures were identified by spectroscopic data. Chiral HPLC analysis indicates the racemic nature of 2–4. Chiral separation followed by X-ray diffraction analysis discloses the absolute configuration of (−)-2. Compounds 1 and 2 could selectively inhibit TGF-β1-induced Smad3 phosphorylation in rat renal tubular epithelial cells, representing novel scaffolds of selective Smad3 activation inhibitors. Total synthesis accompanied by in vivo rodent experiments reveals antifibrotic activities of 1 against kidney fibrosis. Finally, a plausible biosynthetic pathway for 1 was proposed.
1. Introduction
Renal fibrosis, characterized by considerable interstitial myofibroblast activation and excessive matrix protein accumulation, is the downstream event and hallmark of many types of chronic kidney diseases eventually resulting in end stage renal disease.1–3 In recent years, renal fibrosis has become an important topic that attracts broad interest in the field of nephrology.4 Emerging studies have highlighted the importance of partial epithelial to mesenchymal transition, cell cycle arrest and defective cellular metabolism in driving renal fibrosis.4 Among multifaceted factors of renal or other organ fibrogenesis, transforming growth factor β (TGFβ) is generally identified as a central mediator. This pathway has been recognized as a potential target for antifibrotic therapy.5 However, attempts to hinder this process in vivo by neutralizing TGFβ or inhibiting TGFβ receptors is only partially successful.6 Moreover, long-term inhibition of TGFβ/TβRs in experimental animals has led to unwanted diffuse lymphomatous infiltration of several tissues.6 Accumulating evidence has suggested that most fibrogenic activities of TGFβ are mediated through Smad3 rather than Smad2. Thus specific inhibition of Smad3 activation (phosphorylation) by TGFβ leaving Smad2 and non-smad TGFβ pathways intact might be a more useful approach, which could lead to a useful therapy.7 Despite this consensus that scientists in the field reached, specific inhibitors of Smad3 activation are still rare. To the best of our knowledge, SIS3, a synthetic alkaloid as only a tool drug instead of a market product, is one of few examples of specific inhibitors of Smad3 activation.8
Fighting against renal fibrosis has become our research focus. In the course of search for biologically active natural products towards renal fibrosis, GQ5, as a potent and specific p-Smad3 inhibitor, has been characterized by us from Toxicodendron vernicifluum in harmony with its ethnopharmacological knowledge.9 Examination of the backbone of GQ5 reveals that it contains a benzene ring bearing two hydroxyl groups and an aliphatic 15-carbon side chain. Inspired by this observation, lingzhiols, meroterpenoids with similar structure characterization with GQ5, have been identified from Ganoderma lucidum10 and attracted great interest in synthetic chemistry.11–15 In a follow up study on G. lucidum, lingzhifuran A (1) and lingzhilactones D–F (2–4) were isolated (Fig. 1). Compound 1 is characteristic of a complex biosynthetic pathway consisting of a shikimic acid pathway, a mevalonic acid pathway, and an acetic acid pathway. Compounds 2–4 are analogues of lingzhilactones A–C, which were previously isolated from the title species,16 lingzhilactone B thereof has been synthesized by Qin and his co-workers.17 Compounds 1 and 2 were found to be specific Smad3 phosphorylation inhibitors. To examine in vivo effects of 1 against renal fibrosis, subsequent total synthesis followed by biological evaluation in a rodent model were therefore carried out. In this contribution, we describe the isolation and structure characterization of 1–4, total synthesis and in vivo evaluation of 1. In addition, a plausible biosynthetic pathway for 1 is briefly discussed.
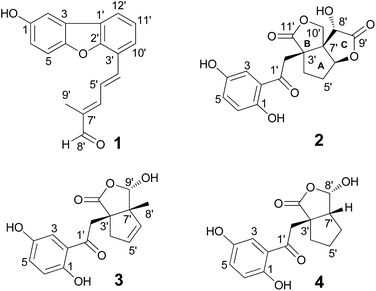 |
| | Fig. 1 The chemical structures of 1–4. | |
2. Results and discussion
2.1. Structure elucidation
Lingzhifuran A (1), has a molecular formula of C18H14O3 derived from its HREIMS (m/z 278.0942 [M]+, calcd for 278.0943), 13C NMR and DEPT spectra, having 12 degrees of unsaturation, the UV spectrum exhibits absorption at 349 nm, implying the presence of a big conjugated system in the molecule. The 1H NMR spectrum (Table 1) shows an aldehyde proton (δH 9.59). The proton signals in the aromatic region are attributed to an ABX system [δH 7.57 (1H, d, J = 8.8 Hz, H-5), 7.08 (1H, dd, J = 8.8, 2.6 Hz, H-6), and 7.52 (1H, d, J = 2.6 Hz, H-2)] and a 1,2,3-trisubstituted benzene ring [δH 7.76 (1H, d, J = 7.6 Hz, H-10′), 7.39 (1H, dd, J = 7.7, 7.6 Hz, H-11′), and 8.01 (1H, d, J = 7.7 Hz, H-12′)]. The 13C NMR and DEPT spectra give 18 carbons classified into one methyl, ten methine (an aldehyde and nine olefinic), seven quaternary carbons (all olefinic, three oxygenated). Apart from one aldehyde group, one benzene ring, five double bonds, the remaining two degrees of unsaturation require the presence of two rings. The 1H-1H COSY spectrum (Fig. 2) give three spin systems which are H-5/H-6, H-10′/H-11′/H-12′, and H-4′/H-5′/H-6′. Two spin systems of them are connected via C-3′, which is supported by the HMBC correlations (Fig. 2) of H-4′, H-5′, H-10′, H-11′/C-3′. The HMBC correlations of H-8′/C-6′, C-7′, C-9′ and H-9′/C-6′, C-7′, C-8′ indicate the position of the methyl and aldehyde groups. The HMBC cross peaks of H-4′, H-10′/C-2′, H-11′, H-12′/C-1′, and H-12′/C-3 suggest that C-12′ is connected to C-1′ allowing the formation of a new benzene ring. The remaining one ring is assumed to be formed by C-4 and C-2′ via an oxygen bridge, in accordance with the chemical shift of C-2′. The large coupling constant of H-4′ (J = 15.6 Hz) indicates that the relationship between H-4′ and H-5′ is E-form. Likewise, ROESY correlations (Fig. 2) of H-6′/H-8′ and H-5′/H3-9′ suggest the trans-relationship of double bond between C-6′ and C-7′. Taken together, the structure of 1 was established as shown.
Table 1 1H and 13C NMR data of 1 and 2 in acetone-d6 (δ in ppm, J in Hz)
| No. |
1 |
No. |
2 |
| δHa |
δCb |
δHc |
δCd |
| 400 MHz. 200 MHz. 600 MHz. 150 MHz. |
| 1 |
|
154.9 |
1 |
|
156.2 |
| 2 |
7.52 (d, 2.6) |
106.9 |
2 |
|
119.4 |
| 3 |
|
125.2 |
3 |
7.38 (d, 2.7) |
115.6 |
| 4 |
|
150.9 |
4 |
|
150.4 |
| 5 |
7.57 (d, 8.8) |
112.9 |
5 |
7.14 (dd, 8.9, 2.7) |
126.7 |
| 6 |
7.08 (dd, 8.8, 2.6) |
116.9 |
6 |
6.84 (d, 8.9) |
119.6 |
| 1′ |
|
126.0 |
1′ |
|
204.5 |
| 2′ |
|
155.3 |
2′a |
3.93 (d, 19.5) |
44.5 |
| 3′ |
|
122.3 |
2′b |
3.58 (d, 19.5) |
|
| 4′ |
7.47 (d, 15.6) |
135.7 |
3′ |
|
53.7 |
| 5′ |
7.92 (dd, 15.6, 11.3) |
127.7 |
4′a |
2.23 (m) |
37.1 |
| 6′ |
7.31 (d, 11.3) |
149.3 |
4′b |
2.08 (m) |
|
| 7′ |
|
139.0 |
5′a |
2.14 (m) |
29.2 |
| 8′ |
9.59 (s) |
194.9 |
5′b |
1.75 (m) |
|
| 9′ |
1.99 (d, 1.2) |
9.7 |
6′ |
5.16 (brd, 3.0) |
93.4 |
| 10′ |
7.76 (d, 7.6) |
127.6 |
7′ |
|
58.4 |
| 11′ |
7.39 (dd, 7.7, 7.6) |
123.9 |
8′ |
4.02 (1H, s) |
71.9 |
| 12′ |
8.01 (d, 7.7) |
122.3 |
9′ |
|
175.0 |
| |
|
|
10′a |
4.92 (d, 9.8) |
70.4 |
| |
|
|
10′b |
4.39 (d, 9.8) |
|
| |
|
|
11′ |
|
180.9 |
| |
|
|
1-OH |
11.08 (s) |
|
| |
|
|
4-OH |
8.32 (brs) |
|
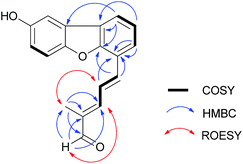 |
| | Fig. 2 Key COSY, HMBC and ROESY correlations of 1. | |
Lingzhilactone D (2) was obtained as a light yellow solid. It has a molecular formula C17H16O8 (10 degrees of unsaturation) derived by analysis of its HREIMS, 13C NMR and DEPT spectra. The 1H NMR spectrum (Table 1) of 2 contains a typical ABX spin system [δH 7.38 (1H, d, J = 2.7 Hz, H-3), 7.14 (1H, dd, J = 8.9, 2.7 Hz, H-5), 6.84 (1H, d, J = 8.9 Hz, H-6)], suggesting the presence of a 1,2,4-trisubstituted benzene ring. The 13C NMR and DEPT spectra contain resonances for 17 carbons including four aliphatic methylene, five methine (two sp3, three sp2), and eight quaternary carbons (one ketone, two carbonyls, three olefinic including two oxygenated, two aliphatic). The proposed presence of a 1,4-dihydroxybenzoyl moiety in 2 is evidenced from HMBC correlations (Fig. 3) of H-3/C-1 (δC 156.2), C-2, C-4 (δC 150.4), C-1′ (δC 204.5), 1-OH (δH 11.08)/C-1. Additional HMBC correlation of H-2′/C-2 (δC 119.4) allows the structure extension of C-1′-C-2′. In the 1H-1H COSY spectrum, correlations of H-4′/H-5′/H-6′ are observed, in conjunct with HMBC correlations of H-4′, H-5′, H-6′/C-3′, C-7′, suggesting the presence of a five-membered lactone ring B, which shares a common C-3′–C-7′ axis with ring A. Furthermore, HMBC correlations of H-2′/C-3′, C-7′, C-11′ indicate that C-2′ is connected to C-3′. In addition to a benzene ring, rings A and B, one ketone and two carbonyls accounting for 9 degrees of unsaturation, the remaining one degree of unsaturation requires an additional ring. The presence of a furolactone ring C is evidenced from HMBC correlations of H-6′/C-8′, C-9′, C-10′, H-8′/C-7′, H-10′/C-8′ and the downfield shift of C-6′ (δC 93.4). As a result, the planar structure of 2 was assigned. The ROESY spectrum of 1 displays correlations between H-8′/Ha-2′, Ha-10′, H-6′/Hb-10′, H-3/Ha-2′, suggesting the relative configurations at ring A as shown in Fig. 3. For the structures like 2, it is challengeable to clarify the stereochemistry at C-8′ of ring C. Chiral analysis by HPLC found that 2 was isolated as a racemic mixture. To assign the absolute configuration at C-8′, subsequent chiral separation followed by single-crystal X-ray diffraction of (−)-2 was fortunately performed, which confirms the relative configurations at ring A and clarifies the absolute configuration at C-8′ (Fig. 4).
 |
| | Fig. 3 Key COSY, HMBC and ROESY correlations of 2. | |
 |
| | Fig. 4 X-ray crystal structure of (−)-2. | |
Lingzhilactone E (3) has the molecular formula C16H16O6 derived by analysis of its HREIMS, 13C NMR and DEPT spectra. The 1H NMR of 3 contains a typical ABX spin system [δH 7.40 (1H, d, J = 2.8 Hz, H-3), 7.12 (1H, dd, J = 8.9, 2.8 Hz, H-5), 6.83 (1H, d, J = 8.9 Hz, H-6)]. The 13C NMR and DEPT spectra (Table 2) exhibit 16 carbons ascribe to one methyl, two sp3 methylene, six methine (five sp2, one oxygenated sp3), seven quaternary carbons (one ketone, one carbonyl, three sp2 including two oxygenated, two sp3). A detailed interpretation of 2D NMR data indicates that the planar structure of 3 is similar to partial structure of 2. 1H-1H COSY correlations of H-4′/H-5′ (δH 5.74)/H-6′ (δH 5.70) indicate a Δ5′,6′ double bond in ring A. HMBC correlations (Fig. 5) of H-9′ (δH 6.00)/C-3′, C-6′, C-7′, C-10′ reveal the presence of ring B and O-atom bearing nature of C-10′. The presence of a methyl attaching to C-7′ in 3 rather than ring C in 2 is evident from HMBC networks of H3-8′/C-3′, C-6′, C-7′, C-9′. The planar structure of 3 was therefore identified. The relative configuration of 3 was assigned by a ROESY experiment which shows correlations of H-2′/H-9′, H-8′, indicating that these protons are spatially adjacent. Chiral HPLC analysis indicates racemic nature of 3. Further preparation by chiral phase on HPLC was not conducted. With these data, the structure of 3 was deduced as shown.
Table 2 1H (600 MHz) and 13C NMR (150 MHz) data of 3 and 4 in acetone-d6 (δ in ppm, J in Hz)
| No. |
3 |
No. |
4 |
| δH |
δC |
δH |
δC |
| 1 |
|
156.4 |
1 |
|
156.4 |
| 2 |
|
119.4 |
2 |
|
119.7 |
| 3 |
7.40 (d, 2.8) |
115.6 |
3 |
7.35 (d, 2.9) |
115.5 |
| 4 |
|
150.3 |
4 |
|
150.3 |
| 5 |
7.12 (dd, 8.9, 2.8) |
126.4 |
5 |
7.11 (dd, 8.9, 2.9) |
126.2 |
| 6 |
6.83 (d, 8.9) |
119.6 |
6 |
6.82 (d, 8.9) |
120.1 |
| 1′ |
|
205.6 |
1′ |
|
205.2 |
| 2′a |
3.74 (d, 19.5) |
43.5 |
2′a |
3.66 (d, 18.8) |
45.0 |
| 2′b |
3.68 (d, 19.5) |
|
2′b |
3.62 (d, 18.8) |
|
| 3′ |
|
54.7 |
3′ |
|
54.5 |
| 4′a |
2.91 (brd, 17.0) |
46.6 |
4′a |
2.01 (overlap) |
39.1 |
| 4′b |
2.71 (brd, 17.0) |
|
4′b |
1.89 (overlap) |
|
| 5′a |
5.74 (dt, 6.0, 1.8) |
128.8 |
5′a |
2.01 (overlap) |
32.8 |
| 6′ |
5.70 (d, 6.0) |
135.3 |
5′b |
1.89 (overlap) |
|
| 7′ |
|
58.7 |
6′a |
1.79 (m) |
25.3 |
| 8′ |
1.09 (s) |
18.0 |
6′b |
1.44 (m) |
|
| 9′ |
6.00 (brs) |
105.3 |
7′ |
2.69 (m) |
53.1 |
| 10′ |
|
180.2 |
8′ |
5.42 (brs) |
104.6 |
| 1-OH |
11.25 (s) |
|
9′ |
|
181.0 |
| 4-OH |
8.34 (brs) |
|
1-OH |
11.28 (s) |
|
| 9′-OH |
6.68 (brs) |
|
4-OH |
8.22 (brs) |
|
| |
|
|
8′-OH |
6.02 (brs) |
|
 |
| | Fig. 5 Key COSY, HMBC and ROESY correlations of 3 and 4. | |
Lingzhilactone F (4) has the molecular formula C15H16O6 derived by analysis of its HREIMS, 13C NMR and DEPT spectra. The 1H and 13C NMR data of 4 resemble those of 3, differing in that the absence of a methyl and a double bond in 4. 1H-1H COSY correlations (Fig. 5) of H-4′/H-5′/H-6′/H-7′/H-8′ strongly support the above conclusion. In the same manner as that of 3, the relative configuration of 4 was deduced by ROESY correlations of H-2′/H-8′, H-7′. Compound 4 was also isolated as a racemate indicated by chiral HPLC analysis. Further preparation by chiral HPLC was not carried out in the present study. Consequently, the structure of 4 was identified as shown.
2.2. Total synthesis of lingzhifuran A (1)
To start with the synthesis of 1, a facile retrosynthetic approach was designed. As shown in Scheme 1, lingzhifuran A (1) could be synthesized from 5 via HWE reaction and deprotection. 5 containing dibenzofuran core could be accessed from 6 through C–H activation of o-halogen bisaryl ether which was developed by Fagnou.18 Intermediate 6 may be formed through condensation by commercially available p-hydroxyanisole and 3-chloro-2-fluoro-benzonitrile.
 |
| | Scheme 1 Retrosynthetic analysis of lingzhifuran A (1). | |
Our synthesis (Scheme 2) began with the preparation of 7. Bisaryl ester 6, which was prepared from p-hydroxyanisole and 3-chloro-2-fluoro-benzonitrile (79%),19 was subjected to a direct intramolecular arylation with aryl chlorides to afford dibenzofuran 7 (86%). While the protecting group of phenol is Me or MOM, it is difficult to acquire pure target molecule due to its poor solubility in several different solvents. In this context, we imagined that silyl group (e.g. TBS) would be appropriate. So we treated 7 with BBr3 to give phenol 8, which was then protected with TBS group to afford 9 (81%, 2 steps). The nitrile of intermediate 9 was converted to aldehyde of intermediate 5 in 77% yield. 5 was subjected to HWE reaction to install the side chain20 of 10 (90%) by reaction with 11, which was prepared from 1-bromo-3-methyl-2-butene.21 Finally, removal of the silyl group afforded 1 (85%). In summary, a concise total synthesis of 1 was achieved in 7 steps with 32% overall yield.
 |
| | Scheme 2 Total synthesis of lingzhifuran A (1). | |
Compounds 1–4 are phenolic meroterpenoids consisting of a hydroquinone and a terpenoidal side chain. Such type of meroterpenoids have been characterized by our group from the genus Ganoderma.16,22,23 The present findings of compounds 1–4 add further facets for Ganoderma chemistry. Compound 1 is characteristic of a dibenzofuran functional group, such kind of moiety is normally found in natural products from lichens and ascomycetes.24 In the plants, dibenzofuran derivatives mainly act as phytoalexins.25 Compound 2, featuring a 5/5/5 polycyclic ring system, is actually a tetranormeroterpenoid, which should be derived from a 15-carbon side chain from a biogenetic point of view. In particular, the formation of ring C in 2 makes this structure unusual.
Analysis of 1 demonstrates that this structure is comprised of an aryl linked a monoterpenoid and a two-carbon unit. With these in hand, we proposed that 1 should be biogenetically derived from a hybridization of the shikimic acid, mevalonic acid and acetic acid pathways (Scheme 3). Briefly, condensation of 4-hydroxybenzonic acid (4HB) and the monoterpene precursor, geranyl diphosphate (GPP), generated geranylhydroxybenzonic acid (A).26 The resulting geranylhydroxybenzonic acid was then oxidized to generate B, a precursor of the reported fornicin D,27 including an oxidative decarboxylation process.28 Homologation of B with pyruvate, which was promoted by pyruvate decarboxylase (PDC), then yielded C.29,30 Cyclization of C afforded D by an intramolecular aldol condensation. Further transformation of D to produce lingzhifuran A (1) via nucleophilic addition, dehydrative elimination, deoxygenation, and oxidation reactions.
 |
| | Scheme 3 A proposed pathway for the biogenesis of 1. | |
2.3. Biological evaluation
2.3.1 Compounds 1 and (+)-2 selectively inhibit TGF-β1–induced Smad3 phosphorylation in vitro. As mentioned above, GQ5 and lingzhiols are potent Smad3 phosphorylation inhibitors. Inspired by the structure relevance between GQ5, lingzhiols, and present isolates, we tend to first examine whether compounds 1–4 could influence TGF-β1–induced activation of Smads pathway in NRK-52E cells. However, only compounds 1 and (+)-2 were tested due to the limited amounts of the isolates. It was found that phosphorylation of Smad2 and Smad3 was significantly activated in the presence of TGF-β1 (Fig. 6, A for 1 and B for (+)-2). Incubation with 1 or (+)-2 could reduce TGF-β1-induced Smad3 rather than Smad2 phosphorylation in a dose-dependent manner. In contrast, the inhibitory effect of 1 or (+)-2 is almost undetectable in NRK-52E cells in the absence of TGF-β1 stimulation even at the concentration of 30 μM. To confirm whether the inhibitory effect of 1 or (+)-2 on Smad3 phosphorylation is specific, other components of the Smads pathway such as Smad4 and Smad7, and other downstream signaling of TGF-β1 were purposely examined. The results show that either 1 or (+)-2 does not affect the protein expression of Smad4 or Smad7 (Fig. 7A and 8A), nor the TGF-β1-induced phosphorylation of p38, ERK, or PI3K (Fig. 7B and 8B). These findings evidently imply that compounds 1 and (+)-2 are both TGF-β1-stimulated Smad3 phosphorylation inhibitors.
 |
| | Fig. 6 Compound 1 (A) or (+)-2 (B) selectively blocks TGF-β1-stimulated Smad3 phosphorylation in a dose dependent manner. NRK-52E cells are treated with TGF-β1 (10 ng mL−1) for 1 h in the absence or presence of different doses of 1 or (+)-2 as indicated. Cell lysates are harvested and immunoblotted with antibodies against p-Smad3, Smad3, p-Smad2 and Smad2. Data are expressed as mean ± SD of three independent experiments. ANOVA, *p < 0.001 versus both TGF-β1 and compound 1 untreated cells. ANOVA, p < 0.01 in p-Smad3 expression in compound 1 treated groups. Ctrl, control. | |
 |
| | Fig. 7 Compound 1 does not affect Smad4 or Smad7 expression (A) or TGF-β1-induced p38, PI3K, ERK phosphorylation (B). NRK-52E cells are treated with TGF-β1 (10 ng mL−1) for 1 h in the absence or presence of 1 at the dose of 30 μM. Cell lysates are harvested and immunoblotted with antibodies against Smad4, Smad7, p-p38, p38, p-PI3K, PI3K, p-ERK and ERK. Data are expressed as mean ± SD of three independent experiments. ANOVA, *p < 0.001 versus both TGF-β1 and compound 1 untreated cells. | |
 |
| | Fig. 8 Compound (+)-2 does not affect Smad4 or Smad7 expression (A) or TGF-β1-induced p38, PI3K, ERK phosphorylation (B). NRK-52E cells ae treated with TGF-β1 (10 ng mL−1) for 1 h in the absence or presence of (+)-2 at the dose of 30 μM. Cell lysates are harvested and immunoblotted with antibodies against Smad4, Smad7, p-p38, p38, p-PI3K, PI3K, p-ERK and ERK. Data are expressed as mean ± SD of three independent experiments. *p < 0.001 versus both TGF-β1 and compound (+)-2 untreated cells. | |
2.3.2 Compounds 1 and (+)-2 inhibit renal fibrosis in vitro. Given that Smad3 phosphorylation mediates fibrogenesis, we next examined whether these two isolates could attenuate renal fibrosis in vitro by checking the alterations of fibrotic markers. As presented in Fig. 9 and 10, compounds 1 or (+)-2 could inhibit TGF-β1-induced α-SMA, collagen I and fibronectin expression at both protein and mRNA levels and in a dose dependent manner, indicating that 1 or (+)-2 could significantly inhibit the downstream gene expression of TGF-β1/Smads signalling and confirming their inhibitory effects on renal fibrosis.
 |
| | Fig. 9 Compound 1 inhibits TGF-β1-induced α-SMA, collagen I and fibronectin expression in a dose dependent manner. (A) NRK-52E cells are treated with TGF-β1 (10 ng mL−1) for 36 h in the absence or presence of different doses of 1 as indicated. Cell lysates are harvested and performed real-time PCR for α-SMA, collagen I and fibronectin. (B) NRK-52E cells are treated with TGF-β1 (10 ng mL−1) for 36 h in the absence or presence of different doses of 1 as indicated. Cell lysates are harvested and immunoblotted with antibodies against α-SMA, collagen I and fibronectin. Data are expressed as mean ± SD of three independent experiments. *p < 0.001 versus both TGF-β1 and compound 1 untreated cells. ANOVA, p < 0.01 in compound 1 treated cells in A and B. Ctrl, control. | |
 |
| | Fig. 10 Compound (+)-2 inhibits TGF-β1-induced α-SMA, collagen I and fibronectin expression in a dose dependent manner. (A) NRK-52E cells are treated with TGF-β1 (10 ng mL−1) for 36 h in the absence or presence of different doses of (+)-2 as indicated. Cell lysates are harvested and performed real-time PCR for α-SMA, collagen I and fibronectin. (B) NRK-52E cells are treated with TGF-β1 (10 ng mL−1) for 36 h in the absence or presence of different doses of (+)-1 as indicated. Cell lysates are harvested and immunoblotted with antibodies against α-SMA, collagen I and fibronectin. Data are expressed as mean ± SD of three independent experiments. *p < 0.001 versus both TGF-β1 and compound (+)-2 untreated cells. ANOVA, p < 0.01 in compound (+)-2 treated cells in A and B. Ctrl, control. | |
2.3.3 Compound 1 inhibits renal fibrosis in vivo. Previous studies show that unilateral ureteral obstruction (UUO) in the rodent could generate progressive renal fibrosis. Thus, ureteral obstruction as a model of renal interstitial fibrosis and obstructive nephropathy has been widely utilized in nephrology.31 Ureteral obstruction causes marked obstructive nephropathy such as inflammatory cell infiltration in the interstitium, tubular degeneration and atrophy, and interstitial fibrosis. The morphologic changes were usually accompanied by expression of α-SMA, collagen, and fibronectin.32,33 To confirm in vivo antifibrotic efficacy of these specific inhibitors on Smad3 phosphorylation, total synthesis of 1 was fulfilled. In light of common administration routes for fibrosis, oral administration for 1 was adopted in this study. The in vivo results show that oral administration of 1 (50 mg kg−1 and 100 mg kg−1) in rats immediately after UUO suppresses renal expression of α-SMA, collagen I, and fibronectin at both mRNA and protein levels (Fig. 11A–D). In addition, late oral administration of 1 (100 mg kg−1) 7 days after UUO also markedly attenuates fibrotic lesions in obstructive nephropathy (Fig. 12A–D), in line with our in vitro study.
 |
| | Fig. 11 Compound 1 dose-dependently attenuates renal interstitial fibrosis in UUO. Rats received daily oral intake of vehicle or 1 at the dose of 50 mg kg−1 day or 100 mg kg−1 day right after UUO, and sacrificed at 14 days. (A) Representative micrographs of HE and Masson trichrome staining demonstrate kidney injury in indicated groups. (B) Real-time PCR analyses for mRNA expression of α-SMA, collagen I and fibronectin in the obstructed kidney. (C) Representative micrographs of the protein expression of α-SMA, collagen I, and fibronectin in the obstructed kidneys. (D) Representative bands (two cases) of western blot analyses for the expression of α-SMA, collagen I and fibronectin in the obstructed kidneys. *p < 0.001 versus sham group in B and D. ANOVA, p < 0.01 in compound 1 treated groups in B and D. n = 6 for each group. | |
 |
| | Fig. 12 Delayed administration of compound 1 attenuates renal interstitial fibrosis in UUO. Rats received daily oral intake of vehicle or 1 at the dose of 100 mg kg−1 day 7 days after UUO, and sacrificed at 14 days. (A) Representative micrographs of HE and Masson trichrome staining demonstrate kidney injury in indicated groups. (B) Real-time PCR analyses for mRNA expression of α-SMA, collagen I and fibronectin in the obstructed kidney. (C) Representative micrographs of the protein expression of α-SMA, collagen I, and fibronectin in the obstructed kidneys. (D) Representative bands (two cases) of western blot analyses for the expression of α-SMA, collagen I and fibronectin in the obstructed kidneys. *p < 0.05 versus sham, #p < 0.05 versus vehicle, n = 6 for each group. | |
3. Experimental section
3.1. General procedure
Column chromatography was performed on silica gel (200–300 mesh; Qingdao Marine Chemical Inc., People's Republic of China), C-18 silica gel (40–60 μm; Daiso Co., Japan), MCI gel CHP 20P (75–150 μm, Mitsubishi Chemical Industries, Tokyo, Japan) and Sephadex LH-20 (Amersham Pharmacia, Sweden). Optical rotations were recorded on a Horiba SEPA-300 polarimeter. UV spectra were recorded on a Shimadzu UV-2401PC spectrometer. CD spectra were measured on a Chirascan instrument. Semi-preparative HPLC was carried out using an Agilent 1200 liquid chromatograph, the column used was a 250 mm × 9.4 mm, i.d., 5 μm, Zorbax SB-C18 and a 250 mm × 4.6 mm, i.d., 5 μm, Daicel Chiralpak IC, flow rate: 1 mL min−1. NMR spectra were recorded on a Bruker AV-400, AV-600 or AV-800 spectrometer, with TMS as an internal standard. EIMS and HREIMS were determined on a AutoSpec Premier P776 spectrometer. ESIMS and HRESIMS were measured on a API QSTAR Pulsar 1 spectrometer. All reagents were commercially available and used without further purification unless indicated otherwise.
3.2. Fungal material
The fruiting bodies of G. lucidum were purchased from the Culture Base of Bei-Zhi-Tang Co., Ltd in Jilin Province, People's Republic of China, in July 2012. A voucher specimen (CHYX-0579) was deposited at the State Key Laboratory of Photochemistry and Plant Resources in West China, Kunming Institute of Botany, Chinese Academy of Sciences, People's Republic of China.
3.3. Extraction and isolation
The dried and powdered G. lucidum (80 kg) was extracted using refluxing with 95% EtOH (2 × 360 L × 2 h) to give a crude extract, which was suspended in water followed by partition with EtOAc to yield an EtOAc soluble extract. The EtOAc extract (1.1 kg) was divided into seven parts (Fr. 1–Fr. 7), by using silica gel column chromatography eluted with a gradient of CHCl3/MeOH (100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1–1:1). Among them, Fr. 2 (55 g) was separated by MCI gel CHP 20P eluted with gradient aqueous MeOH (35–100%) to provide eleven portions (Fr. 2.1–Fr. 2.11). Fr. 2.5 (3.2 g) was filtrated on a Sephadex LH-20 column (MeOH) followed by RP-18 column (MeOH/H2O, 40–60%) to yield eight portions (Fr. 2.5.1–Fr.2.5.8). Fr. 2.5.6 (450 mg) was purified by Sephadex LH-20 (MeOH) followed by semi-preparative HPLC (MeOH/H2O, 65%) to give 1 (2.5 mg). Fr. 3 (170 g) was submitted to a MCI gel CHP 20P column eluted with gradient aqueous MeOH (45–100%) to produce eleven portions (Fr. 3.1–Fr. 3.11). Fr. 3.2 (8.4 g) was passed through Sephadex LH-20 (MeOH) to afford four portions (Fr. 3.2.1–Fr. 3.2.4). Of which Fr. 3.2.4 (350 mg) was subsequently separated by a RP-18 column (MeOH/H2O, 40–70%) and semi-preparative HPLC (MeOH/H2O, 40%) to give 2 (15 mg), 3 (4.5 mg), and 4 (5.5 mg). Compounds 2–4 are racemic mixture analyzed by chiral HPLC. To clarify the absolute configuration of the enantiomers of 2, racemic 2 was subjected to chiral HPLC to yield (+)-2 (5.7 mg) and (−)-2 (5.9 mg) (n-hexane/ethanol, 70:30), respectively.
:1–1:1). Among them, Fr. 2 (55 g) was separated by MCI gel CHP 20P eluted with gradient aqueous MeOH (35–100%) to provide eleven portions (Fr. 2.1–Fr. 2.11). Fr. 2.5 (3.2 g) was filtrated on a Sephadex LH-20 column (MeOH) followed by RP-18 column (MeOH/H2O, 40–60%) to yield eight portions (Fr. 2.5.1–Fr.2.5.8). Fr. 2.5.6 (450 mg) was purified by Sephadex LH-20 (MeOH) followed by semi-preparative HPLC (MeOH/H2O, 65%) to give 1 (2.5 mg). Fr. 3 (170 g) was submitted to a MCI gel CHP 20P column eluted with gradient aqueous MeOH (45–100%) to produce eleven portions (Fr. 3.1–Fr. 3.11). Fr. 3.2 (8.4 g) was passed through Sephadex LH-20 (MeOH) to afford four portions (Fr. 3.2.1–Fr. 3.2.4). Of which Fr. 3.2.4 (350 mg) was subsequently separated by a RP-18 column (MeOH/H2O, 40–70%) and semi-preparative HPLC (MeOH/H2O, 40%) to give 2 (15 mg), 3 (4.5 mg), and 4 (5.5 mg). Compounds 2–4 are racemic mixture analyzed by chiral HPLC. To clarify the absolute configuration of the enantiomers of 2, racemic 2 was subjected to chiral HPLC to yield (+)-2 (5.7 mg) and (−)-2 (5.9 mg) (n-hexane/ethanol, 70:30), respectively.
3.4. Spectral data of the new compounds
3.4.1 Lingzhifuran A (1). Yellow solid; UV (MeOH) λmax (logε) 349 (4.15), 321 (4.22) 256 (4.02), 213 (4.27) nm; ESI-MS m/z 277 [M − H]−; HRESI-MS m/z 278.0942 [M]+ (calcd for C18H14O3, 278.0943); 1H and 13C NMR data, see Table 1.
3.4.2 Lingzhilactone D (2). Yellow solid; {[α]24D +54.2 (c 0.12, MeOH); UV (MeOH) λmax (logε) 370 (3.59), 260 (3.87), 225 (4.15) nm; CD (MeOH) Δε218 − 7.89, Δε375 + 1.23; (+)-2}; {[α]24D −73.2 (c 0.17, MeOH); UV (MeOH) λmax (logε) 370 (3.57), 260 (3.88), 225 (4.11) nm; CD (MeOH) Δε218 + 9.80, Δε375 − 3.85; (−)-2}; ESI-MS m/z 347 [M − H]−; HREIMS m/z 347.0765 [M − H]− (calcd for C17H15O8, 347.0767). 1H and 13C NMR data, see Table 1.
3.4.3 Lingzhilactone E (3). Yellow solid; UV (MeOH) λmax (logε) 369 (3.53), 260 (3.81), 230 (4.05) nm; ESI-MS m/z 303 [M − H]−; 0394 m/z 303.0877 [M − H]− (calcd for C16H15O6, 303.0874). 1H and 13C NMR data, see Table 2.
3.4.4 Lingzhilactone F (4). Yellow solid; UV (MeOH) λmax (logε) 368 (3.70), 259 (3.96), 228 (4.23) nm; ESI-MS m/z 291 [M − H]−; HRESIMS m/z 291.0878 [M − H]− (calcd for C15H15O6, 291.0874); 1H and 13C NMR data, see Table 2.
3.5. Synthesis experimental procedures
3.5.1 3-Chloro-2-(4-methoxyphenoxy)benzonitrile (6). A mixture of p-hydroxyanisole (0.558 g, 4.5 mmol), 3-chloro-2-fluoro-benzonitrile (0.622 g, 4 mmol) and K2CO3 (0.828 g, 6 mmol) in MeCN (25 mL) was refluxed for 8 h. The reaction mixture was cooled when the reaction was completed monitoring by TLC. After removal of MeCN, the reaction mixture was poured into water and extracted with EtOAc (20 mL × 2). The combined organic layer was washed with 3 N aq. NaOH (15 mL) and brine, dried over Na2SO4 and finally removed under reduced pressure. The residue was then purified via silica gel column chromatography (petroleum ether/EtOAc, 20/1) to give 1 (0.820 g, 79%) as a white powder. 1H NMR (400 MHz, CDCl3) δ: 7.69 (dd, J = 8.1, 1.6 Hz, 1H), 7.60 (dd, J = 7.8, 1.6 Hz, 1H), 7.26 (dd, J = 8.1, 7.8 Hz, 1H), 6.83 (m, 4H), 3.78 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 155.6, 153.7, 151.1, 135.6, 132.4, 129.5, 126.0, 116.9, 114.8, 114.7, 109.7, 55.7.
3.5.2 8-Methoxydibenzo[b,d]furan-4-carbonitrile (7). To a two-neck flask containing 6 (0.800 g, 3.1 mmol), Pd(OAc)2 (0.028 g, 0.13 mmol, 4 mol%), PCy3·HBF4 (0.100 g, 0.27 mmol, 8 mol%) and K2CO3 (0.650 g, 4.7 mmol), degassed DMA (20 mL) was added. The resulting suspension was degassed and charged with argon gas followed by heating up to 140 °C and stirring for 15 h. The reaction mixture was cooled when the reaction was completed monitoring by TLC. After cooling down to room temperature, the reaction mixture was poured into water and extracted with EtOAc (30 mL × 3). The combined organic layer was thoroughly washed with water and brine, dried over Na2SO4. Final removal of the solvents under reduced pressure to afford a residue which was purified via silica gel column chromatography (petroleum ether/DCM, 2:1) to give 7 (0.590 g, 86%) as a white powder. 1H NMR (400 MHz, DMSO-d6) δ: 8.51 (d, J = 7.7 Hz, 1H), 8.00 (d, J = 7.6 Hz, 1H), 7.83 (d, J = 2.6 Hz, 1H), 7.77 (d, J = 9.0 Hz, 1H), 7.56 (dd, J = 7.7, 7.6 Hz, 1H), 7.20 (dd, J = 9.0, 2.6 Hz, 1H), 3.89 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ: 156.4, 156.0, 150.2, 131.0, 126.9, 125.3, 123.6, 123.1, 116.9, 115.1, 112.8, 104.8, 95.4, 55.9. EIMS m/z 223 [M]+.
3.5.3 8-Hydroxydibenzo[b,d]furan-4-carbonitrile (8). To a solution of 7 (0.580 g, 2.6 mmol) in DCM (20 mL) at 0 °C, BBr3 (2.5 mL, 5 mmol, 2 M in DCM) was added dropwise. After 30 min at 0 °C, the reaction mixture was allowed to room temperature and stirred for 12 h. The reaction mixture was quenched with water and extracted with EtOAc (50 mL × 3). The EtOAc layer was treated with saturated NaHCO3, brine, dried over Na2SO4 and finally removed under reduced pressure. After evaporation, 8 (0.540 g) was obtained as an off-white solid, which was used for next step without further purification. 1H NMR (400 MHz, acetone-d6) δ: 8.68 (s, 1H), 8.37 (d, J = 7.8, 1.2 Hz, 1H), 7.88 (dd, J = 7.7, 1.2 Hz, 1H), 7.60 (d, J = 8.9 Hz, 1H), 7.57 (d, J = 2.6 Hz, 1H), 7.53 (dd, J = 7.8, 7.7 Hz, 1H), 7.14 (dd, J = 8.9, 2.6 Hz, 1H); 13C NMR (100 MHz, acetone-d6) δ: 157.4, 155.3, 151.2, 131.4, 126.9, 126.6, 124.5, 124.0, 117.9, 115.5, 113.2, 107.2, 97.0; HREIMS m/z 209.0484 [M]+ (calcd for C13H7NO2, 209.0477).
3.5.4 8-((Tert-butyldimethylsilyl)oxy)dibenzo[b,d]furan-4-carbo-nitrile (9). To a solution of 8 (0.520 g, 2.5 mmol) in anhydrous DMF (10 mL) at room temperature, TBSCl (0.450 g, 3 mmol), imidazole (0.340 g, 5 mmol) was added. The reaction mixture was stirred overnight. The reaction mixture was poured into water, extracted with EtOAc (25 mL × 2). The combined organic layer was thoroughly washed with water and brine, dried over Na2SO4 and finally removed under reduced pressure. The residue was separated via silica gel column (petroleum ether/acetone, 15:1) to give 9 (0.650 g, 81%) as a white powder. 1H NMR (400 MHz, CDCl3) δ: 8.11 (dd, J = 7.8, 1.2 Hz, 1H), 7.70 (dd, J = 7.6, 1.2 Hz, 1H), 7.52 (d, J = 8.8 Hz, 1H), 7.39 (dd, J = 7.8, 7.6 Hz, 1H), 7.37 (d, J = 2.6 Hz, 1H), 7.04 (dd, J = 8.8, 2.6 Hz, 1H), 1.03 (s, 9H), 0.24 (s, 6H); 13C NMR (100 MHz, CDCl3) δ: 156.9, 152.3, 151.5, 130.4, 125.8, 125.5, 123.6, 122.7, 121.4, 115.1, 112.6, 111.2, 96.6, 25.7, 18.2, −4.4; HREIMS m/z 323.1337 [M]+ (calcd for C19H21NO2Si, 323.1342).
3.5.5 8-((Tert-butyldimethylsilyl)oxy)dibenzo[b,d]furan-4-carbalde-hyde (5). To a solution of 9 (0.323 g, 1.0 mmol) in anhydrous toluene (20 mL) at 0 °C, DIBALH (1.2 mL, 1.3 mmol, 1.1 M in cyclohexane) was added in a dropwise manner. The reaction mixture was stirred for 15 min at 0 °C when TLC monitoring indicated that only little starting material was left. 20 mL of a saturated solution of Rochelle salt was added. The reaction mixture was stirred for additional 3 h and partitioned with EtOAc (20 mL × 2), which was followed by washing with brine, drying over Na2SO4 and finally removal of solvents under reduced pressure. The residue was then submitted to a silica gel column (petroleum ether/DCM, 3:1) to give 5 (0.252 g, 77%) as a white powder. 1H NMR (400 MHz, CDCl3) δ: 10.58 (s, 1H), 8.15 (d, J = 7.6 Hz, 1H), 7.94 (d, J = 7.7 Hz, 1H), 7.54 (d, J = 8.8 Hz, 1H), 7.45 (dd, J = 7.7, 7.6 Hz, 1H), 7.39 (d, J = 2.6 Hz, 1H), 7.03 (dd, J = 8.8, 2.6 Hz, 1H), 1.03 (s, 9H), 0.25 (s, 6H); 13C NMR (150 MHz, CDCl3) δ: 188.5, 156.7, 152.0, 151.7, 127.5, 126.7, 126.3, 123.5, 122.6, 121.3, 120.9, 112.4, 111.0, 25.7, 18.2, −4.4; HREIMS m/z 326.1325 [M]+ (calcd for C19H22O3Si, 326.1338).
3.5.6 (2E,4E)-5-(8-((Tert-butyldimethylsilyl)oxy)dibenzo[b,d]furan-4-yl)-2-methylpenta-2,4-dienal (10). To a solution of 5 (0.240 g, 0.74 mmol), dimethyl (E)-(3-methyl-4-oxobut-2-en-1-yl)phosphonate (0.170 g, 0.89 mmol) in anhydrous THF (8 mL) at 0 °C, NaH (0.036 g, 0.89 mmol, 60% dispersion in mineral oil) was added in portions. The reaction mixture was quenched with aq. NH4Cl when the reaction halted guided by TLC. The resulting mixture was extracted with EtOAc (15 mL × 2). In the same manner as above, the combined organic layer was washed with brine, dried over Na2SO4 and finally removed under reduced pressure. The resulting residue was passed through silica gel (petroleum ether/EtOAc/DCM, 20:1:2) to yield 10 (0.260 g, 90%) as a yellow powder. 1H NMR (400 MHz, CDCl3) δ: 9.56 (s, 1H), 7.86 (dd, J = 7.6, 1.2 Hz, 1H), 7.77 (dd, J = 15.6, 11.3 Hz, 1H) 7.55 (d, J = 7.5 Hz, 1H), 7.48 (d, J = 8.8 Hz, 1H), 7.37 (d, J = 2.5 Hz, 1H), 7.33 (dd, J = 7.6, 7.5 Hz, 1H), 7.31 (d, J = 15.6 Hz, 1H), 7.11 (d, J = 11.3 Hz, 1H), 7.00 (dd, J = 8.8, 2.5 Hz, 1H), 2.04 (s, 3H), 1.04 (s, 9H), 0.25 (s, 6H); 13C NMR (100 MHz, CDCl3) δ: 194.8, 154.7, 151.8, 151.2, 149.1, 138.2, 135.5, 126.7, 126.6, 125.2, 124.4, 122.8, 121.4, 121.4, 120.3, 112.1, 111.1, 25.7, 18.3, 9.9, −4.4; HREIMS m/z 392.1805 [M]+ (calcd for C24H28O3Si, 392.1808).
3.5.7 (2E,4E)-5-(8-Hydroxydibenzo[b,d]furan-4-yl)-2-methylpenta-2,4-dienal (1). To a solution of 10 (0.250 g, 0.64 mmol) in anhydrous THF (8 mL), TBAF (0.8 mL, 0.8 mmol, 1 M in THF) was added dropwise. The reaction mixture was stirred for 2 h at room temperature, and then quench with 1 N HCl (3 mL). The resulting mixture was adjusted to pH around 4 and extracted with EtOAc. Likewise, the EtOAc layer was washed with brine, dried over Na2SO4 and finally removed under reduced pressure to yield a residue, which was subsequently recrystallized to afford 1 (0.150 g, 85%) as a deep yellow powder. 1H NMR (400 MHz, acetone-d6) δ: 9.59 (s, 1H), 8.46 (br, 1H), 8.02 (dd, J = 7.6, 1.2 Hz, 1H), 7.93 (dd, J = 15.6, 11.3 Hz, 1H), 7.76 (d, J = 7.6 Hz, 1H), 7.58 (d, J = 8.8 Hz, 1H), 7.52 (d, J = 2.6 Hz, 1H), 7.47 (d, J = 15.6 Hz, 1H), 7.39 (dd, J = 7.6, 7.6 Hz, 1H), 7.32 (d, J = 11.3 Hz, 1H), 7.08 (dd, J = 8.8, 2.6 Hz, 1H), 2.00 (d, J = 1.2 Hz, 3H); 13C NMR (150 MHz, acetone-d6) δ: 194.9, 155.3, 154.8, 151.0, 149.3, 139.1, 135.7, 127.7, 127.6, 126.0, 125.2, 123.9, 122.3, 122.3, 116.8, 112.9, 106.8, 9.7. HRESIMS m/z 277.0872 [M − H]− (calcd for C18H13O3, 277.0865). The spectral data are in accordance with those of isolated 1. In addition, the ROSEY correlations at the side chain and HPLC analysis further confirmed the synthetic structure is identical with that of lingzhifuran A.
3.6. Cell culture and treatment
Rat proximal tubular epithelial cells (NRK-52E) were cultured in DMEM-medium supplemented with 10% fetal bovine serum (Gibco, Life Technologies). Compounds were dissolved in DMSO (Sigma) in different concentrations. Cells were treated with compounds at indicated doses for 1 h, followed by incubating with TGF-β1 (10 ng mL−1) for 1 h to examine the effect of phosphorylation of Smad3, Smad2, p38, PI3K and ERK, and to examine the effect of protein expression of Smad4 and Smad7. Cells were collected for Western blot analyses. To test the antifibrotic action of compounds, cells were treated with compounds at indicated doses for 1 h, followed by incubating with TGF-β1 (10 ng mL−1) for 36 h for real-time PCR analyses and 48 h for Western blot analyses.
3.7. Real-time PCR
Total RNA was prepared from the NRK-52E cells using TRIzol reagent (Invitrogen, Carlsbad, CA, USA), according to the manufacturer's instructions (Invitrogen, Carlsbad, CA, USA). RNA concentration was calculated using a Nanodrop ND1000 spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA). Aliquots of each RNA extraction were reverse-transcribed simultaneously into cDNA using One-Step RT-PCR kit (TaKaRa, Tokyo, Japan) according to the manufacturer's protocol. Each quantitative real-time PCR was performed in a total volume of 25 μL in duplicate by using the SYBR®Premix Ex Taq™ kit (TaKaRa, Kyoto, Japan) and the Fast Real-Time PCR system 7500 (Applied Biosystems Inc., Foster City, CA, USA). The sequences of the primer pairs are as follows: rat collagen I: forward 5′-TGCCGTGACCTCAAGATGTG-3′, reverse 5′-CACAAGCGTGCTGTAGGTGA-3’; rat α-SMA: forward 5′-GATCACCATCGGGAATGAACGC-3’; reverse 5′-CTTAGAAGCATTTGCGGTGGAC-3’; rat fibronectin: forward 5′- CGAAACCATGAACTTTCTGC -3′, reverse 5′- CCTCAGTGGGCACACACTCC-3’; rat GAPDH: forward 5′-TCCGCCCCTTCCGCTGATG-3′, reverse 5′-CACGGAAGGCCATGCCAGTGA-3′. The thermal cycling conditions comprised a 30 seconds step at 95 °C, followed by 95 °C for 5 seconds and 60 °C for 34 seconds for 40 cycles with melting curve analysis. Relative quantification of each gene was calculated after normalization to GAPDH mRNA by using the 2-ΔΔCT method.
3.8. Western blot analyses
Western blot analyses for specific protein expression were performed essentially according to an established procedure.34 The primary antibodies used were as follows: anti α-SMA (Sigma), anti collagen I (catalog number 234167, Calbiochem), anti fibronectin (catalog number F6348, Sigma); anti p-Smad2 (catalog number 3108S), anti Smad2 (catalog number 5339S), anti p-Smad3 (catalog number 9520S), anti Smad3 (catalog number 9523S), anti Smad4 (catalog number 9515), anti Smad7 (Sigma), anti p-p38 (catalog number 9211S), anti p38 (catalog number 9212), anti p-PI3K (catalog number 4228S), anti PI3K (catalog number 4292), anti p-ERK (catalog number 4377S) and anti ERK (catalog number 4695, Cell Signaling Technology, Inc., Beverly, MA).
3.9. Animal model and treatment
3.9.1 Ethics statement. This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol was approved by the Committee on the Ethics of Animal Experiments of the Nanfang Hospital.Male Sprague-Dawley rats with body weight 200 to 250 g were purchased from the Animal Experiment Committee of the Southern Medical University and housed in the Nanfang Hospital Animal Center. UUO was performed using an established protocol as described.35 To evaluate the effect of compound 1 on renal fibrosis, the rats were randomized into four groups (n = 6 in each group): (1) sham operated mice; (2) UUO rats received daily oral intake of vehicle; (3) UUO rats treated with daily oral intake of 1 50 mg kg−1 day; (4) UUO rats treated with daily oral intake of 1 100 mg kg−1 day. Compound 1 was suspended in PBS. All the mice were sacrificed 14 days after UUO. To evaluate the effect of delay administration of 1 on renal fibrosis, the rats were randomized into three groups (n = 6 in each group): (1) sham operated rats; (2) UUO rats received daily oral intake of vehicle; (3) UUO rats treated with daily oral intake of 1 100 mg kg−1 day 7 days after UUO. All the rats were sacrificed 14 days after UUO.
3.10. Morphological and immunohistochemical analyses
Two micrometer sections of paraffin-embedded kidney tissue were subjected to Masson Trichrome or HE staining using the commercial kits (Sigma, St. Louis, MO, USA) according to the manufacturer's protocols. Immunohistochemical staining was performed on 4 μm kidney sections as previously described.36 Briefly, the kidney sections were stained by anti-α-SMA (Sigma, St. Louis, MO, USA), anti-collagen I (Calbiochem, San Diago, USA), anti-fibronectin (Sigma, St. Louis, MO, USA), and detected by the Evision/HRP Kit (Dako, CA, USA).
4. Conclusions
In conclusion, we isolated four new meroterpenoids from G. lucidum. X-ray diffraction analysis clarified the absolute configuration of (−)-2. Compounds 1 and (+)-2 were identified as potent specific inhibitors of TGF-β1-induced Smad3 phosphorylation and show in vitro activity against renal fibrosis. Total synthesis of 1 was accomplished and in vivo antifibrotic data are in line with the in vitro results. Further work on 1 such as structure modification, action mechanism exploration is required.
Acknowledgements
This study was supported by NSFC-Joint Foundation of Yunnan Province (U1202222), National Science Fund for Distinguished Young Scholars (81525026), National Natural Science Foundation of China (21472199), and a project from State Key Laboratory of Phytochemistry and Plant Resources in West China, Kunming Institute of Botany (P2016-ZZ02).
Notes and references
- M. Zeisberg and E. G. Neilson, J. Am. Soc. Nephrol., 2010, 21, 1819 CrossRef CAS PubMed.
- M. Nangaku, Intern. Med., 2004, 43, 9 CrossRef CAS PubMed.
- Y. Liu, Kidney Int., 2006, 69, 213 CrossRef CAS PubMed.
- D. Zhou and Y. Liu, Nat. Rev. Nephrol., 2016, 12, 68 CrossRef CAS PubMed.
- X. M. Meng, P. M. K. Tang, J. Li and H. Y. Lan, Front. Physiol., 2015, 6, 82 Search PubMed.
- W. Zhang, M. Tsuda, G.-X. Yang, K. Tsuneyama, X. S. He, A. A. Ansari, W. M. Ridgway, R. L. Coppel, Z.-X. Lian, P. S. C. Leung and M. E. Gershwin, PLoS One, 2012, 7, e49413 CAS.
- H. Y. Lan, Kidney Res. Clin. Pract., 2012, 31, 4 CrossRef PubMed.
- M. Jinnin, H. Ihn and K. Tamaki, Mol. Pharmacol., 2006, 69, 597 CrossRef CAS PubMed.
- J. B. He, J. Luo, L. Zhang, Y. M. Yan and Y. X. Cheng, Org. Lett., 2013, 15, 3602 CrossRef CAS PubMed.
- Y. M. Yan, J. Ai, L. L. Zhou, A. C. K. Chung, R. Li, J. Nie, P. Fang, X. L. Wang, J. Luo, Q. Hu, F. F. Hou and Y. X. Cheng, Org. Lett., 2013, 15, 5488 CrossRef CAS PubMed.
- R. Long, J. Huang, W. Shao, S. Liu, Y. Lan, J. Gong and Z. Yang, Nat. Commun., 2014, 5, 5707 CrossRef CAS PubMed.
- D. Chen, H. M. Liu, M. M. Li, Y. M. Yan, W. D. Xu, X. N. Li, Y. X. Cheng and H. B. Qin, Chem. Commun., 2015, 51, 14594 RSC.
- K. Sharmah Gautam and V. B. Birman, Org. Lett., 2016, 18, 1499 CrossRef CAS PubMed.
- X. Li, X. Liu, X. Jiao, H. Yang, Y. Yao and P. Xie, Org. Lett., 2016, 18, 1944 CrossRef CAS PubMed.
- D. Chen, W. D. Xu, H. M. Liu, M. M. Li, Y. M. Yan, X. N. Li, Y. Li, Y. X. Cheng and H. B. Qin, Chem. Commun., 2016, 52, 8561 RSC.
- Y. M. Yan, X. L. Wang, L. L. Zhou, F. J. Zhou, R. Li, Y. Tian, Z. L. Zuo, P. Fang, A. C. K. Chung, F. F. Hou and Y. X. Cheng, J. Ethnopharmacol., 2015, 176, 385 CrossRef CAS PubMed.
- D. Chen, X. M. Li, H. M. Liu, M. M. Li, Y. X. Cheng and H. B. Qin, Tetrahedron Lett., 2016, 57, 2877 CrossRef CAS.
- L. C. Campeau, M. Parisien, A. Jean and K. Fagnou, J. Am. Chem. Soc., 2006, 128, 581 CrossRef CAS PubMed.
- H. Kelgtermans, L. Dobrzańska, L. V. Meervelt and W. Dehaen, Org. Lett., 2012, 14, 1500 CrossRef CAS PubMed.
- A. Mohamed Hachi, E. About Jaudet, J. C. Combret and N. Collignon, Synthesis, 1999, 1999, 1188 CrossRef.
- L. D. Li, W. Tang, W. J. Liu and Z. B. Zhao, Youji Huaxue, 2008, 28, 489 CAS.
- M. Dou, L. Di, L. L. Zhou, Y. M. Yan, X. L. Wang, F. J. Zhou, Z. L. Yang, R. T. Li, F. F. Hou and Y. X. Cheng, Org. Lett., 2014, 16, 6064 CrossRef CAS PubMed.
- F. J. Zhou, Y. Nian, Y. Yan, Y. Gong, Q. Luo, Y. Zhang, B. Hou, Z. L. Zuo, S. M. Wang, H. H. Jiang, J. Yang and Y. X. Cheng, Org. Lett., 2015, 17, 3082 CrossRef CAS PubMed.
- M. Millot, A. Dieu and S. Tomasi, Nat. Prod. Rep., 2016, 33, 801 RSC.
- T. Kokubun and J. B. Harborne, Phytochemistry, 1995, 40, 1649 CrossRef CAS.
- Q. Luo, L. Tian, L. Di, Y. M. Yan, X. Y. Wei, X. F. Wang and Y. X. Cheng, Org. Lett., 2015, 17, 1565 CrossRef CAS PubMed.
- X. Peng, J. Liu, C. Wang, Z. Han, Y. Shu, X. Li, L. Zhou and M. Qiu, Food Chem., 2015, 171, 251 CrossRef CAS PubMed.
- Y. L. Yang, H. Zhou, G. Du, K. N. Feng, T. Feng, X. L. Fu, J. K. Liu and Y. Zeng, Angew. Chem., Int. Ed., 2016, 55, 5463 CrossRef CAS PubMed.
- D. Hansson, A. Menkis, Å. Olson, J. Stenlid, A. Broberg and M. Karlsson, Phytochemistry, 2012, 84, 31 CrossRef CAS PubMed.
- P. J. Silk and J. B. Macaulay, FEMS Microbiol. Lett., 2003, 228, 11 CrossRef CAS PubMed.
- R. L. Chevalier, M. S. Forbes and B. A. Thornhill, Kidney Int., 2009, 75, 1145 CrossRef PubMed.
- S. Klahr and J. Morrissey, Am. J. Physiol. Renal Physiol., 2002, 283, F861 CrossRef PubMed.
- J. A. Moon, H. T. Kim, I. S. Cho, Y. Y. Sheen and D. K. Kim, Kidney Int., 2006, 70, 1234 CrossRef CAS PubMed.
- J. H. Li, H. J. Zhu, X. R. Huang, K. N. Lai, R. J. Johnson and H. Y. Lan, J. Am. Soc. Nephrol., 2002, 13, 1464 CrossRef CAS PubMed.
- J. Yang, C. Dai and Y. Liu, J. Am. Soc. Nephrol., 2002, 13, 2464 CrossRef CAS PubMed.
- K. H. Yoo, B. A. Thornhill, M. S. Forbes, C. M. Coleman, E. S. Marcinko, L. Liaw and R. L. Chevalier, Kidney Int., 2006, 70, 1735 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: 1D, 2D NMR, and MS spectra, crystallographic data. CCDC 1491784. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra17900b |
| ‡ These authors contributed equally. |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 














