
C54Si6 heterofullerene as a potential gas sensor for CO, NO, and HCN detection†
Yongliang Yong*ab,
Shijie Lva,
Ruizhou Zhanga,
Qingxiao Zhoua,
Xiangying Sua,
Tongwei Lia and
Hongling Cuia
aCollege of Physics and Engineering, Henan University of Science and Technology, Luoyang 471003, China. E-mail: ylyong@haust.edu.cn; Tel: +86-187-36385204
bDepartment of Physics, Zhejiang University, Hangzhou 310027, China
First published on 13th September 2016
Abstract
The adsorption of CO, NO, and HCN molecules on the C54Si6 heterofullerene is investigated on the basis of density functional theory calculations to exploit its potential applications as a gas sensor. The C54Si6 heterofullerene has two highly stable isomers (named isomer-1 and isomer-2). We find that the toxic CO, NO, and HCN molecules are chemically adsorbed on isomer-1 with moderate adsorption energies and apparent charge transfer. The electronic properties of isomer-1 are significantly influenced by the CO, NO, and HCN adsorption, especially its electric conductivity. The recovery time of the isomer-1 sensor for CO, NO, and HCN at room temperature is estimated to be short due to the medium (optimal) adsorption energies, indicating that isomer-1 (i.e. the most stable configuration) of C54Si6 heterofullerene should be a good CO, NO, and HCN sensor. Similar analysis indicates that the isomer-2 of C54Si6 heterofullerene is a potential efficient gas sensor for NO detection.
1. Introduction
Since the detection of biological and chemical species in the atmosphere or process gases is of great concern in relation to environmental pollution, industrial emission monitoring and process control, medical diagnosis, public security, agriculture and a variety of industries, much research has been focused on the development of novel chemical sensors with superior performance.1–3 In recent years, nanoclusters have become promising candidates for gas sensing applications because of their excellent properties.4–14 For example, one of the characteristics of nanoclusters is their high surface area–volume ratio. Gas sensors involve a surface response process, so a high surface area–volume ratio favors the adsorption of gases on the sensor and can accelerate charge accumulation, improving the sensitivity and response velocity.Carbon monoxide (CO), nitrogen monoxide (NO) and hydrogen cyanide (HCN) are the ones of the most dangerous and commonest gases in air pollution and for human beings and animals. They are highly toxic and extremely dangerous because it is colorless and odorless. CO can be produced by the incomplete combustion of fuels and is commonly found in automobile exhaust, the burning of domestic fuels, and so on. The NO and HCN gases, formed during combustion processes in power plants, waste incinerators and combustion engines such as automobiles, is among the major air pollutants leading to the formation of photochemical smog and acid rain. The U.S. Environmental Protection Agency (EPA) recommends an ambient air quality of 9 ppm CO or lower averaged over 8 h and 35 ppm or lower over 1 h. At CO levels above 70 ppm, symptoms can include headache, fatigue and nausea. At sustained CO concentrations above 150–200 ppm, disorientation, unconsciousness, and death are possible. In addition, exposure levels of 100 ppm HCN which would result in death is about 1 hour or less in some cases, while exposure levels of 500 ppm HCN is within 15 minutes. Higher concentration levels will result in faster onset of symptoms or death. CO, NO and HCN sensors are, therefore, required in various situations, including the detection of smoldering fires and air quality in urban and closed environments.
The discovery of C60 fullerene15 meant a breakthrough in the cluster field and inaugurated the research line on carbon-based nanotechnology. For example, the adsorption of DNA on C60 fullerene has been widely studied experimentally and theoretically for the purpose of C60-based electrochemical biosensors, which has recently been reviewed by Pilehvar et al.16 However, common gas molecules, such as CO, NO, CO2, and H2, are all physically adsorbed on the C60 fullerene with very small adsorption energies, and can hardly influence the electronic properties of C60 fullerene,17,18 which prevents immediate practical applications, for example as a molecule sensor. Recently, the C60-based heterofullerenes are found that they can expand potential applications in developing of catalysts, gas sensors, gas storage, and electronic devices,19–27 since they combine the remarkable structural stability of pure fullerenes with the enhanced chemical reactivity acquired via replacement, encapsulation, or coating by heteroatoms. For example, Zahedi et al. have studied the adsorption of NH3 and NO2 molecules on C48B6N6 and C30B15N15 heterofullerenes, respectively.23,24 They found that the C48B6N6 heterofullerene is a suitable sensor material for NO2 detection, while the C48B6N6 and C30B15N15 heterofullerenes are ideal materials for elimination and filtering of ammonia. Baei et al. have predicted that C30B15N15 heterofullerene can be viewed as a potential electronic sensor for NO detection.25 Hassani et al.27 have investigated the geometries, stabilities and electronic properties of simple and iodine adsorbed S-doped fullerenes using density functional theory (DFT) calculations, and found that the electrical conductivity of the S-doped fullerenes was increased upon the iodine adsorption and the S-doped fullerenes could be used in sensor devices for iodine detection.
In recently years, heterofullerenes based on C60 containing the element of silicon have been investigated extensively.28–35 As known, silicon shares the same column of the periodic table with carbon, and doping fullerene with silicon does not alter the valence electrons, which is different from other substitutions such as B and N. Thus the doping of silicon may be friendly to the pristine fullerene, which brings new properties without breaking the innate structure and stability. Besides, the electronic structure of the Si-doped fullerene would likely to be similar to that of pristine fullerene from viewpoint of total occupancy of the energy levels. On the other hand, silicon strongly prefers the sp3-like bonding, which contrasts with the chemical nature of carbon. This makes it capable of interacting via single, double and triple bonds with itself and other atoms, indicating that the chemical reactivity of fullerene is more inclined to increase due to the substitutional Si-doping. Geometry optimization within the framework of DFT provides clear evidence of stable fullerene-like cage structures for C60−mSim (m ≤ 30).28–33 Mass spectrometry and photofragmentation experiments suggest that up to 12 Si atoms can be substituted in a C60 fullerene without destroying its structure.34,35 Most recently, the adsorption properties of H2 molecules on Si-doped C60 heterofullerenes have been studied in order to develop their potential applications for hydrogen storage.36 However, to our knowledge, little attention has been focused on molecules adsorption on Si-doped C60 heterofullerenes to investigate their potential applications for gas sensors. Therefore, it would be interesting and important to find out that whether the Si-doped C60 heterofullerenes would be suitable as gas sensors to detect a certain toxic gas. Among the Si-doped C60 heterofullerenes, two most stable configurations of C54Si6 heterofullerene are found to be thermal stable under the temperature of 2000 K.28 In the present work, the adsorption behaviors and electronic properties of CO, NO and HCN on C54Si6 heterofullerene are studied via first-principles calculations, to explore the possibility of C54Si6 heterofullerene as gas sensors to detect CO, NO and HCN gases.
2. Computational methods
All the geometry optimizations and energy calculations are performed using DMOL3 program package (Accelyrs Inc.)37,38 at the level of density functional theory. The gradient-corrected (GGA) density functionals for exchange (Becke39) and correlation (Perdew–Wang40) along with double numeric basis sets supplemented with d polarization functions (i.e., the DNP set) are used here. The all-electron treatment is chosen to describe all atoms in a given system. Self-consistent field procedures are performed with a convergence criterion of 10−6 a.u. on the energy and electron density. The geometries are fully optimized without any symmetry constraints. We use a convergence criterion of 10−3 a.u. on the gradient and displacement and 10−5 a.u. on the total energy in geometrical optimization. Normal-mode vibrational analysis is applied to make sure that the obtained structures are real local minima. The charge transfer between C54Si6 heterofullerene and absorbed molecules is analyzed based on Hirshfeld analysis, which is based directly on the electron density as a function of space.In this paper, the adsorption energies (Eads) are defined as Eads = E(complex) − E(heterofullerene) − E(molecule), where E(complex), E(heterofullerene), and E(molecule) stand for the total energies of the molecule–heterofullerene complex, the pristine heterofullerene, and the corresponding molecule, respectively. The accuracy of the GGA-BP and DNP combination for investigating the structures and properties of the system, which includes Si, C, N, and B atoms, has been confirmed by the previous studies.41–43
3. Results and discussion
C54Si6 heterofullerene can be obtained via substitutionally doping six Si atoms in C60 fullerene. The two most stable configurations of C54Si6 heterofullerene are shown in Fig. 1. The most stable configuration (denoted as isomer-1) consists of four identical Si–Si ph (pentagon–hexagon) bonds (2.343 Å) with two sets of equal Si–C distances (1.879 Å and 1.826 Å), which is shown in Fig. 1A. The second stable isomer as shown in Fig. 1B (denoted as isomer-2) has all Si atoms on a hexagon, and the difference energy between the two isomers is only 0.121 eV. The most stable structure exhibits a gap of 0.871 eV between the highest occupied (HOMO) and the lowest unoccupied (LUMO) molecular orbitals, and the second stable isomer has a HOMO–LUMO gap of 1.081 eV. These results we obtained are essentially in agreement with previous reports.28,33 Previous studies28,33 have reported that the other isomers of C54Si6 heterofullerene are much less stable than the two most stable configurations. Because of the small energy difference between the two stable configurations, we then consider the adsorption of CO, NO and HCN on the two isomers of C54Si6 heterofullerene. As mentioned above, the pure C60 fullerene is less reactive with chemical species such as gas molecules, however, doping with heteroatoms can efficiently tailor the structures and physicochemical properties of C60, silicon has four electrons in its valence shell and prefers to bind with sp3 hybridization, but the introduction of the Si-dopants into C60 would make the Si atoms bond with an sp2 type structure, thus creating a located state. Furthermore, as we shall describe later, most states of HOMOs of the Si-doped C60 are localized around the Si-dopants, indicating that the Si atoms have much higher reactivity than other atoms. Thus, the Si-dopants act as the active sites for foreign adsorbates, as will be tested by the following results. | ||
| Fig. 1 The two most stable structures of C54Si6. Values in parentheses are relative energies in eV. The distances are in Angstrom. Here and in the following figures, yellow balls represent Si atoms, and gray balls represent C atoms. | ||
To confirm the most favorable site for the considered molecule adsorption on C54Si6 heterofullerene, we first investigate all possible geometries with the molecules attached to the heterofullerene. Since the chemical reactivity of Si units is much stronger than that of C units, we mainly considered that the molecule (CO, NO, and HCN) was initially placed on the sites near the Si units, but the representative sites near C units, such as the sites of top of a C atom, the center of a six-membered (or five-membered) ring, are also considered. The final optimized configurations of molecule–heterofullerene complexes are shown in Fig. 2–6, and results of our calculations are summarized in Tables 1 and 2.
 | ||
| Fig. 2 Two most stable configurations of the CO molecule adsorbed on the isomer-1 of C54Si6. Red balls represent O atoms. Values in parentheses are relative energies in eV. Bond distances are in Angstrom. | ||
| Systema | Eads (eV) | Eg (eV) | ET (e) |
|---|---|---|---|
| a Notations: e.g. CO–A represents the optimized stable structure of CO molecule adsorbed on the isomer-1 of C54Si6 as shown in the corresponding figure (A). | |||
| CO–A | −0.265 | 0.703 | 0.168 |
| CO–B | −0.138 | 0.677 | −0.088 |
| NO–A | −0.721 | 0.306 | −0.546 |
| NO–B | −0.666 | 0.253 | −0.087 |
| NO–C | −0.447 | 0.332 | −0.606 |
| HCN–A | −0.854 | 0.587 | −0.189 |
| HCN–B | −0.390 | 0.755 | 0.311 |
Then we first investigate the adsorption of CO molecule on the isomer-1 of C54Si6 heterofullerene. After full relaxation of all possible initial geometries, the CO molecule adopts the orientation of C atom pointing to the heterofullerene, and the configuration of CO located on the top of Si atoms is the most stable, as shown in Fig. 2A, which is similar to the case of CO molecule adsorption on silicon carbide nanotubes.44 At this configuration the adsorption energy is −0.265 eV, and the distance between molecule and heterofullerene is 2.000 Å. This C–Si distance is consistent with the cases of SinCn clusters.41,42 Our calculations demonstrate the CO molecule can be adsorbed on the top of each Si atom as the same form as the most stable one. There is an unambiguous charge transfer of 0.168 e from the CO molecule to the heterofullerene. The second stable configuration as shown in Fig. 2B is found that the CO molecule is located on the center of C–C bond with the adsorption energy of −0.138 eV, and charge transfer of 0.088 e from the heterofullerene to the CO molecule. It should be noted that although the Hirshfeld scheme gives much smaller atomic charges than nearly all other charge schemes,45 it is much more reliable than Mulliken, Bader, and Weinhold schemes.46 It can be seen from Table 1 that the HOMO–LUMO gap of the two stable structures of CO–heterofullerene complexes is 0.703 and 0.677 eV, respectively. Comparing the HOMO–LUMO gap of the CO–heterofullerene complexes with the pristine heterofullerene (0.871 eV) clarifies significant influences of CO adsorption on the heterofullerene electronic properties. Therefore, both configurations of CO adsorption on the heterofullerene can be characterized by the moderate adsorption energies, obvious charge transfer between CO molecule and the heterofullerene, and the change of HOMO–LUMO gap.
For the NO molecule adsorbed on the isomer-1 of the heterofullerene, it is found from Fig. 3 that, differing from the case of CO–heterofullerene complexes, the NO molecule is energetically more feasible for the adsorption on the heterofullerene with the N–O bond parallel to Si–Si bond. At the most stable configuration, the adsorption energy is −0.721 eV, and the distance of Si–N and Si–O bonds are 1.760 and 1.701 Å, respectively. It is clear from Fig. 3 that the structure of C54Si6 changes too much after adsorbing NO, when the molecule is parallel to the heterofullerene. Meanwhile, the N–O bond length of NO molecule is expanded from 1.165 Å in molecular form to about 1.50 Å. Moreover, the orientation of N–O bond pointing to Si–Si bond influences the stability of the NO–heterofullerene complexes. However, the adsorption of NO, which is similar to the case of the most stable configuration of CO adsorption, does not lead to structural deformation of heterofullerene (see Fig. 3B). This configuration has an adsorption energy of −0.666 eV, which is a little higher in energy by 0.055 eV than the most stable one. The charge analysis using the Hirshfeld method indicates that in the configurations of NO parallel to the heterofullerene, a net charge (more than 0.5 e) is transferred from the heterofullerene to the NO molecule, while in the configurations of NO perpendicular to the heterofullerene a net charge of 0.087 e is transferred from the heterofullerene to the NO molecule. It can be seen from Table 1 that the three most stable configurations have HOMO–LUMO gaps of 0.306, 0.253, and 0.332 eV, respectively, which is much smaller than that of pure heterofullerene (0.871 eV), indicating that the electronic properties of the heterofullerene have dramatically changed after NO adsorption.
 | ||
| Fig. 3 Three most stable configurations of the NO molecule adsorbed on the isomer-1 of C54Si6. Blue balls represent N atoms. Values in parentheses are relative energies in eV. Bond distances are in Angstrom. | ||
The interactions between the isomer-1 of the C54Si6 heterofullerene and HCN molecule are also investigated in the present work. First, the possible structures of HCN–heterofullerene complexes are explored. The structures are optimized, and the most and second stable configurations are shown in Fig. 4. Similar to the case of NO molecule adsorption on the isomer-1 of the C54Si6 heterofullerene, the N and C atom in HCN molecule are attached to Si atoms with N–C bond parallel to Si–Si bond, and the N–Si and C–Si distance is 1.820 and 1.973 Å, respectively. At this configuration the adsorption energy is −0.854 eV, and the charge transfer from the heterofullerene to the HCN molecule is 0.189 e. The HCN molecule adopts the orientation of N atom pointing to the heterofullerene, and the configuration of HCN located on top of one Si atom with the molecule perpendicular to the heterofullerene surface is less stable than the most stable one, which has an adsorption energy of −0.390 eV, higher by 0.464 eV than that of the most stable one. However, at this configuration, there is a net charge of 0.311 e from the HCN molecule to heterofullerene. The HOMO–LUMO gap of the two complexes is 0.587 and 0.755 eV, respectively, smaller than that of the pure heterofullerene.
 | ||
| Fig. 4 Two most stable configurations of the HCN molecule adsorbed on the isomer-1 of C54Si6. White balls represent H atoms. Values in parentheses are relative energies in eV. Bond distances are in Angstrom. | ||
We next discuss the adsorption of the three molecules on the isomer-2 of the C54Si6 heterofullerene. The adsorption of NO molecule on the isomer-2 of the C54Si6 heterofullerene is firstly considered. As shown in Fig. 5, an NO molecule can be chemically adsorbed on the Si atoms in isomer-2 of the heterofullerene. In the most stable configuration, the N and O atom in NO molecule approach Si atoms with an adsorption energy of −0.717 eV. The adsorption energy of the second stable one is 0.356 eV. These adsorption energies are consistent with the results of NO molecule adsorbed on the isomer-1 of the heterofullerene. Furthermore, the adsorption of NO makes the NO–heterofullerene complexes have an obvious charge transfer between the molecule and heterofullerene and much smaller HOMO–LUMO gaps (less than 0.42 eV) than that of the pure heterofullerene (1.081 eV) (see Table 2).
 | ||
| Fig. 5 Two most stable configurations of the NO molecule adsorbed on the isomer-2 of C54Si6. Values in parentheses are relative energies in eV. Bond distances are in Angstrom. | ||
The adsorption of CO on the isomer-2 of the C54Si6 heterofullerene is similar to the case of CO adsorption on the isomer-1, and the CO molecule can approach each Si atom in the heterofullerene with adsorption energies of about −0.365 eV. After adsorption, the charge transfer from the CO molecule to the heterofullerene is 0.364 e, however, the HOMO–LUMO gap of the CO–heterofullerene complex is 1.031 eV, which is a little smaller than that of pristine heterofullerene, indicating the adsorption of CO does not significantly influence the heterofullerene electronic properties. From Fig. 6, we can see that the HCN molecule can only be physically adsorbed on the isomer-2 of the heterofullerene with the adsorption energies less than −0.09 eV. The heterofullerene–molecule distance is larger than 3.20 Å, and there is no apparent charge transfer between the molecule and the heterofullerene. This indicates that the adsorption is physically electrostatic and it is not strong enough to present desorption at room temperature. It is noted that all the above discussed configurations are found to have no imaginary frequencies, indicating that they are real stable.
 | ||
| Fig. 6 The most stable configurations of (A) CO and (B) HCN molecule adsorbed on the isomer-2 of C54Si6. Values in parentheses are relative energies in eV. Bond distances are in Angstrom. | ||
It is well known that van der Waals (vdW) forces play an important role in adsorption processes, and vdW correction is not represented well by the standard DFT-GGA functional. In order to consider the influence of the vdW interaction between C54Si6 heterofullerene and molecules, the dispersion-corrected DFT (DFT-D) based on Tkatchenko–Scheffler methods47 are chosen. We only considered the most stable configurations of molecules adsorbed on C54Si6 heterofullerene. The configurations, adsorption energy, and adsorption distance are shown in Fig. S1 (see ESI†). It is found that the configurations with considering the vdW interaction do not have obvious structural deformation with respect to those without considering the vdW interaction. Further, after considering the vdW forces, the change of the adsorption energies for the most stable configurations is very small (in the range of 0.032–0.089 eV). This results indicate that the dispersion forces have a little influence in the interaction between C54Si6 heterofullerene and (CO, NO, and HCN) molecules. Thus, we believe that the standard DFT-GGA functional would be reliable to describe the interaction between C54Si6 heterofullerene and (CO, NO, and HCN) molecules.
It is noted that for the three (CO, NO and HCN) molecules adsorbed on the isomer-1 of C54Si6, the adsorption energy is much lower for HCN, however, there is a marginal difference between CO and NO. This may be because of the difference of bond energies between the bonds of Si–O, Si–N, Si–C and Si–Si. It is known that the strengths of bonds become weaker and weaker with the order of Si–O (452 kJ mol−1), Si–N (355 kJ mol−1), Si–C (318 kJ mol−1) and Si–Si (222 kJ mol−1) bonds.48 It can be seen from Fig. 2–4 that the Si–C and Si–N bonds are formed for HCN molecule adsorbed on the C54Si6, while the Si–O and Si–N bonds are formed for NO on C54Si6, however, there is only one Si–C bond formed for CO on C54Si6. Therefore, the adsorption energies of HCN and NO on C54Si6 are much lower than that of CO on C54Si6. However, it is noted that the adsorption of NO on C54Si6 make the N–O bond of NO expand from 1.165 Å in molecular form to about 1.50 Å, which would weaken significantly the strength of N–O bond. On the contrary, the lengths of C–N and C–H bonds in HCN do not change remarkably before and after the adsorption, indicating that the strengths of C–N and C–H bonds do not change remarkably. This may explain why the adsorption energy of HCN on the isomer-1 is the lowest.
We next discuss the potential application of the C54Si6 heterofullerene for gases detection based on our results. It is well known that, for a certain kind of gases detection, the gas molecule should be chemically adsorbed on C54Si6 heterofullerene with moderate adsorption energy, and has conspicuous charge transfers between molecule and the heterofullerene to influence the electrical conductivity of the clusters. Our results indicate that CO, NO, and HCN molecules can be chemically adsorbed on the isomer-1 of the C54Si6 heterofullerene with proper adsorption energies (≥0.26 eV), which are large enough to restrain spontaneous desorption at room temperature. In addition, the adsorption of CO, NO, and HCN molecules leads to an obvious charge transfer (≥0.168 e) between the molecule and the isomer-1 of C54Si6 heterofullerene. These results make the isomer-1 of C54Si6 heterofullerene suitable for CO, NO, and HCN detection. However, the isomer-2 of C54Si6 heterofullerene is suitable for CO and NO detection. Furthermore, we consider the influences of the adsorption of the three molecules on electronic properties of C54Si6 heterofullerene. Fig. 7 shows the total and partial density of states (DOS) of the two isomers of the C54Si6 heterofullerene and the most stable structures of C54Si6 with gas molecule adsorption. For the most stable configurations of CO and HCN molecules adsorbed on the isomer-2 of the C54Si6 heterofullerene, it can be found that the DOSs near the Fermi level are not influenced by the CO and HCN adsorption (Fig. 7f and h). Thus the HOMO–LUMO gap (Eg) of the isomer-2 of C54Si6 (1.081 eV) has no crucial change due to the CO and HCN adsorption (the change of Eg is 0.05 and 0.007 eV for the CO and HCN adsorption, respectively). However, for the other cases, it can be seen from Fig. 7b–d and g that because of the molecule adsorption some energy states appear above the Fermi level so that the Eg has significant change. The change of the HOMO–LUMO gaps for the configurations of CO, NO, and HCN molecules adsorbed on the isomer-1 of C54Si6 heterofullerene is in the range of 0.168 to 0.565 eV (19.2% to 64.9%), indicating high sensitivity of electronic properties of the isomer-1 of C54Si6 heterofullerene toward the three molecules adsorption. However, our results indicate that only the adsorption of NO can obviously change the electronic properties of the isomer-2 of C54Si6 heterofullerene (see Table 2). The changes in HOMO–LUMO gaps (Eg) of the configurations can reflect the electric conductivity change of C54Si6 heterofullerene according to the formula,49  , where σ is the electric conductivity of the configurations, k is the Boltzmann's constant, and T is the thermodynamic temperature. By this definition, a smaller Eg value at a given temperature results in a larger electric conductivity. Therefore, the CO, NO, and HCN molecules can be detected by calculating the conductivity change of the isomer-1 of C54Si6 heterofullerene before and after the adsorption process. Meanwhile, the NO molecule also can be detected by calculating the conductivity change of the isomer-2 of C54Si6 heterofullerene before and after the adsorption process.
, where σ is the electric conductivity of the configurations, k is the Boltzmann's constant, and T is the thermodynamic temperature. By this definition, a smaller Eg value at a given temperature results in a larger electric conductivity. Therefore, the CO, NO, and HCN molecules can be detected by calculating the conductivity change of the isomer-1 of C54Si6 heterofullerene before and after the adsorption process. Meanwhile, the NO molecule also can be detected by calculating the conductivity change of the isomer-2 of C54Si6 heterofullerene before and after the adsorption process.
 | ||
| Fig. 7 Total and partial density of states (DOS) for the two isomers of the C54Si6 heterofullerene and the most stable structures of C54Si6 with gas molecule adsorption. (a) The isomer-1 of C54Si6, (b) CO, (c) NO, (d) HCN adsorbed on the isomer-1 of C54Si6, (e) the isomer-2 of C54Si6, (f) CO, (g) NO, (h) HCN adsorbed on the isomer-2 of C54Si6. The vertical line indicates the Fermi level. | ||
As it is mentioned, after calculating and comparing the adsorption configurations of molecules on C54Si6, we found that the two isomers of C54Si6 exhibit different adsorption properties to the molecules (CO, NO, and HCN). For better understand of these observations, we have performed frontier molecular orbital analysis on the molecules, the two isomers of C54Si6, and the molecule–heterofullerene complexes, which is shown in Fig. 8 and 9. It is seen that in the CO and HCN molecules, the σ state shape HOMO is mainly distributed around the C atom while the π shape LUMO appears around all the atoms, however, in the NO molecule, the π shape HOMO and LUMO appear around both atoms. The LUMO is mainly distributed throughout the Si–C bonds in the isomer-1 of C54Si6 while the LUMO in the isomer-2 of C54Si6 is mainly distributed around the Si atoms. Therefore, the adsorption sites are always located on the Si atoms neighboring the C atoms in the in the isomer-1 of C54Si6 (or the Si-hexagon of the isomer-2). Furthermore, it can be seen from Fig. 9 that after the CO and NO molecules adsorption on the isomer-2 of C54Si6, significant changes are observed in HOMO and LUMO, indicating that the electron density are redistributed. This indicates that the isomer-2 of C54Si6 have high reactivity to CO and NO molecules. The same conclusion has been found in the cases of the three molecules (CO, NO, and HCN) adsorbed on the isomer-1 of C54Si6. However, after the HCN molecule adsorbed on isomer-2 of C54Si6, no redistribution of charge density is observed as the LUMO of the HCN-adsorbed system is still the LUMO of the isomer-2 of C54Si6, thereby reflecting the absence of any covalent interaction between the two. In addition, for the isomer-2 of C54Si6, this structure has a larger HOMO–LUMO gap (1.081 eV) than that of the isomer-1, indicating a stronger electronic stability. This may further indicate that the isomer-2 of C54Si6 is not easy to interact with other molecules.
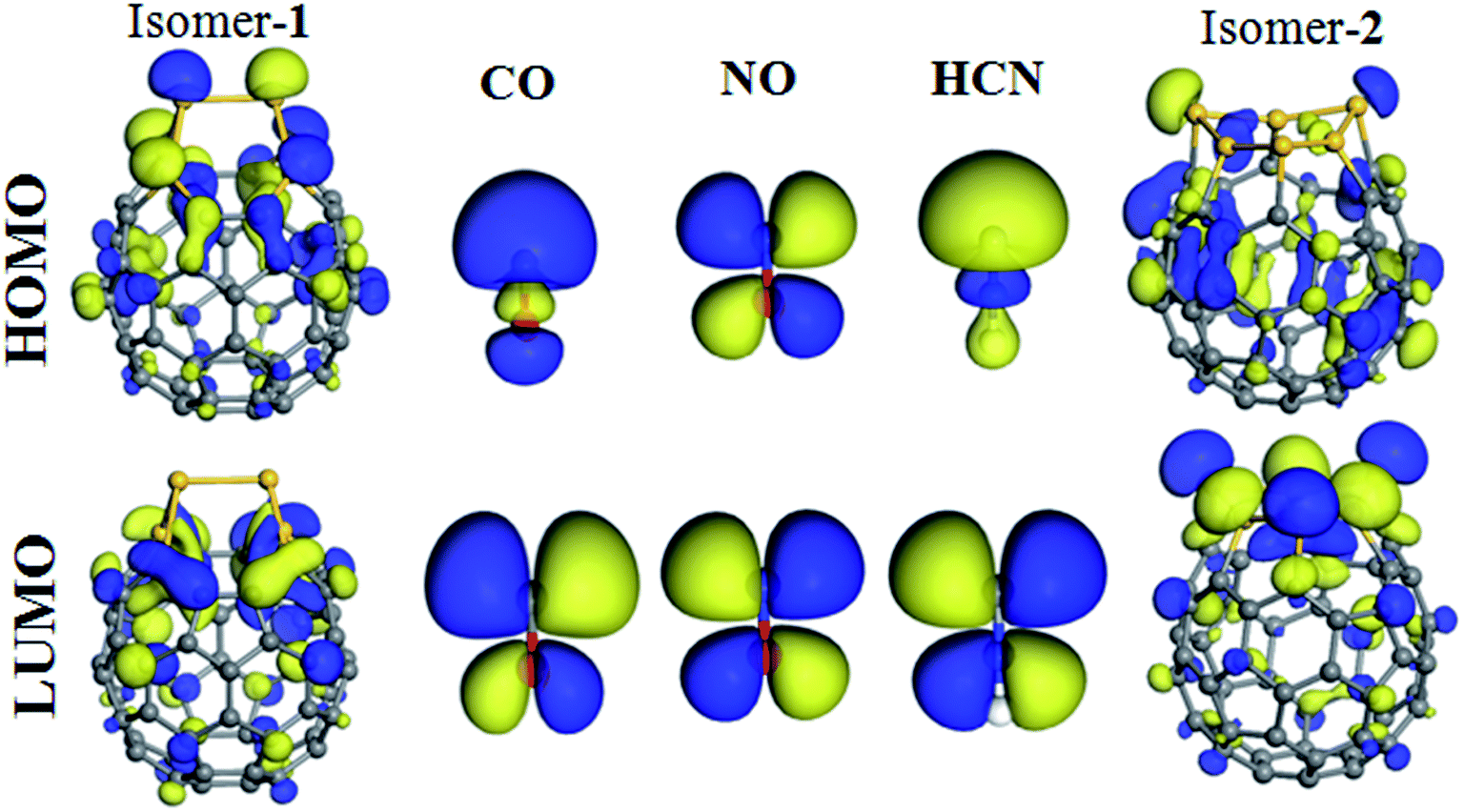 | ||
| Fig. 8 The HOMO and LUMO pictures of the two isomers of the C54Si6 heterofullerene and the CO, NO, and HCN molecules. | ||
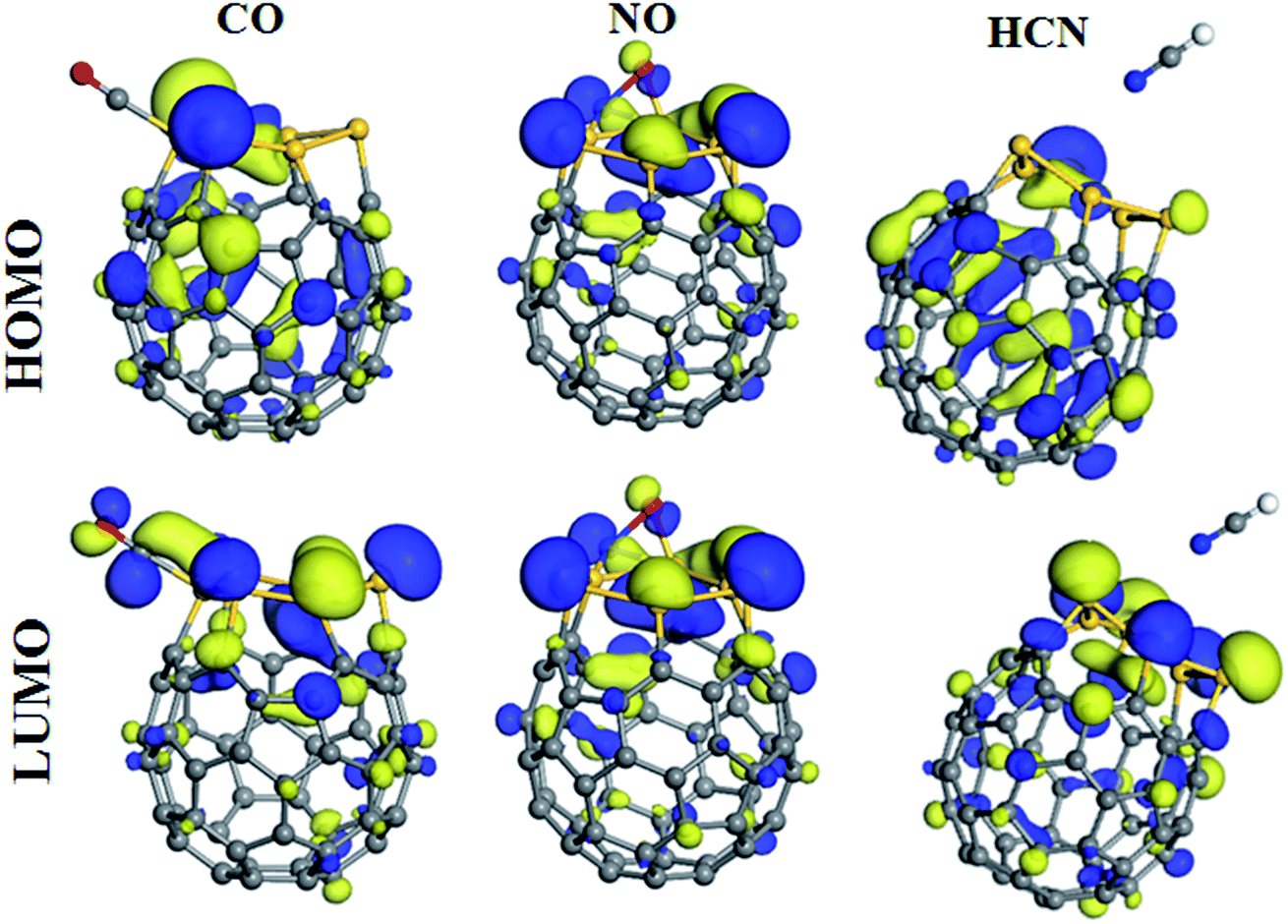 | ||
| Fig. 9 The HOMO and LUMO pictures of the most stable configurations of the CO, NO, and HCN molecules adsorbed on the isomer-2 of the C54Si6 heterofullerene. | ||
Furthermore, it worth noting that the strong interaction between the heterofullerene and certain molecules may also brings some serious drawbacks. That is to say, strong adsorption of a certain molecule on the C54Si6 heterofullerene implies that desorption of this gas molecule from the C54Si6 heterofullerene could be quite difficult and the devices may suffer from a longer recovery time. The recovery time τ, which relates to the adsorption energy Eads, can be expressed as τ = ν0−1e−Eads/kT, where ν0 the attempt frequency, k the Boltzmann's constant, and T the temperature. Based on this formula, Peng et al.50 predicted that the recovery time of carbon nanotubes-based sensors for NO2 at room temperature is in the range of 5 μs and 16 s for the adsorption energies of −0.34 to −0.79 eV, and the recovery time of 12 h corresponds to adsorption energy of −1.00 eV. Note that the adsorption energies of CO, NO, and HCN molecules on the C54Si6 heterofullerene are moderate (−0.265 to 0.854 eV), the recovery time of the C54Si6 heterofullerene sensor for CO, NO, and HCN at room temperature may be short, indicating that it is possible to desorb them from C54Si6 heterofullerene by heating at room temperature. Therefore, the isomer-1 of C54Si6 heterofullerene should be good CO, NO, and HCN sensors with quick response as well as short recovery time, while the isomer-2 of C54Si6 heterofullerene should be good NO sensors. We hope that our theoretical predictions will inspire the interest of future experimental researchers into the applications of the C54Si6 heterofullerene. Since both the heterofullerene and most of polymers can be viewed as large molecules, the structure and operation mechanism of the C54Si6 heterofullerene based gas sensors should be similar to the case of polymer-based gas sensors. Bartlett's group has presented a basic model for polymer gas sensors as early as 1994,51 which may be applied to heterofullerene based sensors. Some deposition techniques such as thermal evaporation and Langmuir–Blodgett technique, which have been successfully used to polymer gas sensors,52 can be developed to realize the sensing layer of C54Si6 heterofullerene.
4. Conclusions
In summary, the adsorption of CO, NO, and HCN molecules on the C54Si6 heterofullerene are explored on the basis of density functional theory calculations. The C54Si6 heterofullerene has two highly stable isomers (named isomer-1 and isomer-2). We find that the toxic molecules (CO, NO, and HCN) are chemically adsorbed on the isomer-1 of C54Si6 heterofullerene with moderate adsorption energies, and can lead to finite charge transfer. The electronic properties of isomer-1 of C54Si6 heterofullerene are significantly influenced by the CO, NO, and HCN adsorption, especially their electric conductivity. Meanwhile, the electronic properties of isomer-2 of C54Si6 heterofullerene are significantly influenced only by NO adsorption. The moderate adsorption energies of CO, NO, and HCN molecules on C54Si6 heterofullerene make the recovery time of the C54Si6 heterofullerene sensor for CO, NO, and HCN at room temperature short, indicating that the C54Si6 heterofullerene should be good CO, NO, and HCN sensors with quick response and short recovery time. Therefore, the most stable configuration (i.e. isomer-1) of C54Si6 heterofullerene can be expected to be an excellent gas sensor for CO, NO, and HCN detection, while the isomer-2 (the second most stable one) of C54Si6 heterofullerene is a potential candidate for a molecule sensor with high sensitivity for NO.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 11304080 and No. 51302065) and the Innovation Team of Henan University of Science and Technology (No. 2015XTD001).References
- L. E. Kreno, K. Leong, O. K. Farha, M. Allendorf, R. P. van Duyne and J. T. Hupp, Chem. Rev., 2012, 112, 1105–1125 CrossRef CAS PubMed.
- G. Jiménez-Cadena, J. Riu and F. X. Rius, Analyst, 2012, 132, 1083–1099 RSC.
- G. F. Fine, L. M. Cavanagh, A. Afonja and R. Binions, Sensors, 2010, 10, 5469–5502 CrossRef CAS PubMed.
- A. W. Castleman and P. Jena, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 10554–10559 CrossRef CAS PubMed.
- R. Ferrando, J. Jellinek and R. L. Johnston, Chem. Rev., 2008, 108, 845–910 CrossRef CAS PubMed.
- T. Zhao, T. Zhou, Q. Yao, C. Hao and X. Chen, J. Environ. Sci. Health, Part C: Environ. Carcinog. Ecotoxicol. Rev., 2015, 33, 168–187 CrossRef CAS PubMed.
- Y. Yong, C. Li, X. Li, T. Li, H. Cui and S. Lv, J. Phys. Chem. C, 2015, 119, 7534–7540 CAS.
- A. Mathew and T. Pradeep, Part. Part. Syst. Charact., 2014, 31, 1017–1053 CrossRef CAS.
- H. Haick, J. Phys. D: Appl. Phys., 2007, 40, 7173–7186 CrossRef CAS.
- J. van Lith, A. Lassesson, S. A. Brown, M. Schulze, J. G. Partridge and A. Ayesh, Appl. Phys. Lett., 2007, 91, 181910 CrossRef.
- Y. Yong, X. Li, T. Li, H. Cui and S. Lv, Europhys. Lett., 2015, 111, 10006 CrossRef.
- K. Suematsu, Y. Shin, Z. Hua, K. Yoshida, M. Yuasa, T. Kida and K. Shimanoe, ACS Appl. Mater. Interfaces, 2014, 6, 5319–53269 CAS.
- A. Soltani, S. G. Raz, M. R. Taghartapeh, A. V. Moradi and R. Z. Mehrabian, Comput. Mater. Sci., 2013, 79, 795–803 CrossRef CAS.
- J. Beheshtian, A. A. Peyghan and Z. Bagheri, Comput. Mater. Sci., 2012, 62, 71–74 CrossRef CAS.
- H. W. Kroto, J. R. Heath, S. C. O'brien, R. F. Curl and R. E. Smalley, Nature, 1985, 318, 162–163 CrossRef CAS.
- S. Pilehvar and K. D. Wael, Biosensors, 2015, 5, 712–735 CrossRef PubMed.
- O. Echt, A. Kaiser, S. Zöttl, A. Mauracher, S. Denifl and P. Scheier, ChemPlusChem, 2013, 78, 910–920 CrossRef CAS.
- S. M. Gatica, M. M. Calbi, R. D. Diehl and M. W. Cole, J. Low Temp. Phys., 2008, 152, 89–107 CrossRef CAS.
- J. C. Hummelen, B. Knight, J. Pavlovich, R. González and F. Wudl, Science, 1995, 269, 1554–1556 CAS.
- P. Trogadas, T. F. Fuller and P. Strasser, Carbon, 2014, 75, 5–42 CrossRef CAS.
- L. Dai, D. W. Chang, J. Baek and W. Lu, Small, 2012, 8, 1130–1166 CrossRef CAS PubMed.
- M. D. Tzirakis and M. Orfanopoulos, Chem. Rev., 2013, 113, 5262–5321 CrossRef CAS PubMed.
- E. Zahedi and A. Seif, Phys. B, 2011, 406, 3704–3709 CrossRef CAS.
- E. Zahedi, A. Seif and T. S. Ahmadi, J. Comput. Theor. Nanosci., 2011, 8, 2159–2165 CrossRef CAS.
- M. T. Baei, K. Tavakoli, S. Hashemian and P. Torabi, Fullerenes, Nanotubes, Carbon Nanostruct., 2015, 23, 153–157 CrossRef CAS.
- Q. Li, J. Zheng, J. Dang and X. Zhao, ChemPhysChem, 2015, 16, 390–395 CrossRef CAS PubMed.
- F. Hassani and H. Tavakol, Sens. Actuators, B, 2014, 196, 624–630 CrossRef CAS.
- M. Matsubara and C. Massobrio, J. Chem. Phys., 2005, 122, 084304 CrossRef CAS PubMed.
- P. A. Marcos, J. A. Alonso and M. J. López, J. Chem. Phys., 2005, 123, 204323 CrossRef CAS PubMed.
- M. Matsubara, J. Kortus, J. C. Parlebas and C. Massobrio, Phys. Rev. Lett., 2006, 96, 155502 CrossRef CAS PubMed.
- M. Matsubara, C. Massobrio and J. Parlebas, Comput. Mater. Sci., 2005, 33, 237–243 CrossRef CAS.
- M. Matsubara and C. Massobrio, J. Phys. Chem. A, 2005, 109, 4415–4418 CrossRef CAS PubMed.
- L. Koponen, M. J. Puska and R. M. Nieminen, J. Chem. Phys., 2008, 128, 154307 CrossRef PubMed.
- M. Pellarin, C. Ray, J. Lermé, J. L. Vialle, M. Broyer, X. Blase, P. Kéghélian, P. Mélinon and A. Perez, J. Chem. Phys., 1999, 110, 6927–6938 CrossRef CAS.
- X. Blase, M. Broyer, P. Kéghélian, J. Lermé, P. Mélinon, M. Pellarin, A. Perez, C. Ray and J. L. Vialle, Eur. Phys. J. D, 1999, 9, 49–54 CrossRef.
- M. D. Ganji, N. Ahmadian, M. Goodarzi and H. A. Khorrami, J. Comput. Theor. Nanosci., 2011, 8, 1392–1399 CrossRef CAS.
- B. Delley, J. Chem. Phys., 1990, 92, 508–517 CrossRef CAS.
- B. Delley, J. Chem. Phys., 2000, 113, 7756–7764 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1988, 88, 2547–2553 CrossRef CAS.
- J. P. Perdew and Y. Wang, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 45, 13244–13249 CrossRef.
- J. Hou and B. Song, J. Chem. Phys., 2008, 128, 154304 CrossRef PubMed.
- B. Song, Y. Yong, J. Hou and P. He, Eur. Phys. J. D, 2010, 59, 399–406 CrossRef CAS.
- Y. Yong, B. Song, J. Li and P. He, J. Phys. B: At., Mol. Opt. Phys., 2011, 44, 135101 CrossRef.
- R. Q. Wu, M. Yang, Y. H. Lu, Y. P. Feng, Z. G. Huang and Q. Wu, J. Phys. Chem. C, 2008, 112, 15985–15988 CAS.
- E. R. Davidson and S. Chakravorty, Theor. Chim. Acta, 1992, 83, 319–1330 CrossRef CAS.
- C. F. Guerra, J.-W. Handgraaf, E. J. Baerends and F. M. Bickelhaupt, J. Comput. Chem., 2004, 25, 189–210 CrossRef CAS PubMed.
- A. Tkatchenko and M. Scheffler, Phys. Rev. Lett., 2009, 102, 073005 CrossRef PubMed.
- T. L. Cottrell, The Strengths of Chemical Bonds, Butterworths, London, 2nd edn, 1958 Search PubMed.
- S. Li, Semiconductor Physical Electronics, Springer, USA, 2nd edn, 2006 Search PubMed.
- S. Peng, K. Cho, P. Qi and H. Dai, Chem. Phys. Lett., 2004, 387, 271–276 CrossRef CAS.
- J. W. Gardner and P. N. Bartlett, Synth. Met., 1994, 57, 2665–2670 Search PubMed.
- H. Bai and G. Shi, Sensors, 2007, 7, 267–307 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Configurations, adsorption energies and adsorption distances of the CO, NO, and HCN molecules adsorbed on C54Si6 heterofullerene with considering van der Waals interaction. See DOI: 10.1039/c6ra17834k. |
| This journal is © The Royal Society of Chemistry 2016 |