
Green method to fabricate porous microspheres for ultrasensitive SERS detection using UV light†
Abstract
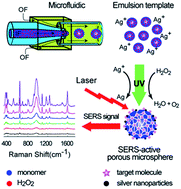
A green method was developed to fabricate a novel porous polymer microsphere with SERS activity, which can be used for ultrasensitive detection because of its preconcentration function. Ultraviolet irradiation as “green” reduction was successfully achieved through several complex processes, namely, monomer solidification, hydrogen peroxide photolysis, and silver nanoparticle fabrication, in only one step. During the solidification process, H2O and O2, which are originally from H2O2 reduction, escaped from the emulsion templates, resulting in various interconnected micro-/nano-channels. The mean pore size of the prepared porous microspheres was 4 μm, and the specific surface area can reach 31 m2 g−1. The silver nanoparticles produced in the reduction process of silver nitrate were deposited on the porous surface of the microsphere and contributed to stable SERS activity with a SERS enhancement factor of up to 3.8 × 106 and a low detection limit of 10−13 M level for rhodamine 6G. This green method provides highly sensitive and uniform SERS substrates and ensures the stability and reliability of our “SERS substrate”.
 Please wait while we load your content...
Please wait while we load your content...

