
Capacity control of ferric coordination polymers by zinc nitrate for lithium-ion batteries†
Xiaobing
Lou,  ‡a
Ming
Shen,
*a
‡a
Ming
Shen,
*a
‡a
Ming
Shen,
*a
Abstract
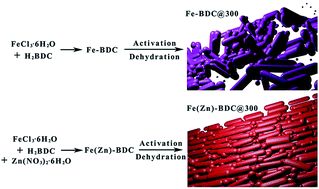
Ferric coordination polymers were synthesized via a hydrothermal process. With the addition of zinc nitrate, the as-prepared Fe(Zn)–BDC@300 shows rod-like morphology and exhibits superior electrochemical performance as an anode material for lithium-ion batteries. It shows a reversible capacity of 863.4 mA h g−1 at 0.1 A g−1 after 120 cycles.
 Please wait while we load your content...
Please wait while we load your content...

