
ZnII and CdII MOFs based on an amidoisophthalic acid ligand: synthesis, structure and catalytic application in transesterification†
Anirban Karmakar *,
Guilherme M. D. M. Rúbio,
M. Fátima C. Guedes da Silva*,
Ana P. C. Ribeiro and
Armando J. L. Pombeiro*
*,
Guilherme M. D. M. Rúbio,
M. Fátima C. Guedes da Silva*,
Ana P. C. Ribeiro and
Armando J. L. Pombeiro*
Centro de Química Estrutural, Instituto Superior Técnico, Universidade de Lisboa, Av. Rovisco Pais, 1049-001, Lisbon, Portugal. E-mail: anirbanchem@gmail.com; fatima.guedes@tecnico.ulisboa.pt; pombeiro@tecnico.ulisboa.pt
First published on 12th September 2016
Abstract
Solvothermal reaction of zinc(II) and cadmium(II) salts with 5-propionamidoisophthalic acid (H2L1) and 5-benzamidoisophthalic acid (H2L2) in the presence or absence of an auxiliary ligand gives rise to a series of 1D, 2D and 3D Zn(II) or Cd(II) self-assembled metal–organic frameworks as revealed by X-ray diffraction structural analyses: [Zn(μ-L1-μ-1κOO′:2κO′′)(formamide)2]n (1), [{Zn2(μ-L1-1κO:2κO′)2(4,4′-bipyridine)2(H2O)}·2(DMF)·5(H2O)]n (2), [Cd(μ-L1-μ-1κO2O′:2κO′′2O′′′)(DMF)]n (3), [{Zn(μ-L2-1κO:2κO′)(4,4′-bipyridine)(H2O)}·(H2O)]n (4) and [{Cd(μ-L2-μ-1κO2O′:2κO′′2O′′′)(formamide)(4,4′-bipyridine)}·(formamide)]n (5), which are also characterized by elemental analysis, IR spectroscopy and thermogravimetric analysis. The different architectures found in 1–5 result from the coordination modes of the carboxylate groups, which can assume nonbridging monodentate (in 1, 2 and 4), bridging bidentate (also in 1), bridging chelate (in 3 and 5) and chelate bidentate (also in 5) coordination. While 1 and 5 possess one dimensional double chain type structures, 3 has a 1D cyclic type structure and 2 and 4 have 2D or 3D wave-like structures, respectively. Topological analysis has shown that 1 and 5 have a 3-connected uninodal net with a topological type SP1-periodic net, 2 has a 2,4-connected binodal net, 3 has a 4-connected uninodal net structure and 4 has a more complex 2,2,2,4-connected tetranodal net. Frameworks 1–5 act as heterogeneous catalysts for the transesterification reaction of different carboxylate esters, with 4 showing the highest activity. These heterogeneous catalysts can be recycled without losing activity.
Introduction
Metal–organic frameworks (MOFs) are crystalline coordination networks consisting of metal ions or clusters and multidentate organic ligands.1 This research field is undergoing rapid growth, not only because of the intriguing architectures of MOFs but also due to potential applications as functional materials in catalysis, magnetism, nonlinear optics, gas storage and separation, etc.2 Their crystalline nature, large surface area, high porosity, tuneable surface properties, adequate framework stability and the presence of active sites either in the framework or hosted in the voids also make MOFs very promising as heterogeneous catalysts for liquid phase reactions.3However, the controlled synthesis of MOFs is still a challenge on account of the many factors that influence the product structures, such as the nature of the ligand and metal, solvent, reaction temperature, time, metal to ligand ratio, pH and method of crystallization etc.4 In particular, multidentate carboxylate ligands are good candidates for the construction of novel structures and topologies since they have diverse coordination modes and can adopt different conformations. Rigid benzene dicarboxylate,5 tricarboxylate,6 tetracarboxylate7 and azolate-based ligands,8 as well as their derivatives, are commonly used as organic building blocks, and this approach has effectively produced a wide range of MOFs with interesting properties.
MOFs constructed from amide based linkers have attracted considerable attention due to their ability to provide guest-accessible functional organic sites and to obtain controlled architectures with interesting topologies.9 Recently, we have focused on the design of various types of coordination polymers and discrete complexes using amido carboxylate ligands and different metal ions and on the study of their catalytic activity in various organic transformations.10 The amide based ligands are chosen because they can construct kinetically inert and thermodynamically stable metal complexes and the presence of the amide group in the framework provides an extra opportunity for guest binding. Besides, the presence of carboxylate groups allows slight conformational changes for the potential assembly of different network architectures.
Heterogeneous catalysis concerns one of the earliest proposed applications for crystalline MOFs and presently many organic reactions are effectively catalyzed by them.11 Recently, our group has reported various MOFs which are catalytically active for the oxidation,12a–c nitroaldol,10,12d cyanosilylation12e and Knoevenagel condensation reactions.12f Now we wish to extend our study to another type of reaction, i.e., transesterification, which concerns an important transformation in organic synthesis, in industrial and in academic laboratories,13 with applications in polyester synthesis and biodiesel production.14 Many homogenous catalysts have been developed for these reactions,15 but usually they require high reaction temperature and acidic reaction conditions. Lately, some MOFs have been used to catalyze such a type of reactions,16 but there is still a demand to develop new types of catalysts based on cheap and environmentally tolerable compounds that could be easily recyclable and show a high efficiency under mild conditions.
Thus, the main objectives of the present work are: (i) to synthesize Zn(II) and Cd(II)-MOFs by using 5-propionamidoisophthalic acid (H2L1) and 5-benzamidoisophthalic acid (H2L2) linkers in the presence or absence of auxiliary ligands, under various solvothermal conditions; (ii) to apply the synthesized MOFs as heterogeneous catalysts for the transesterification reaction.
Hence, we report herein the solvothermal synthesis and characterization of five new self-assembled zinc(II) and cadmium(II) MOFs, namely [[Zn(μ-L1-μ-1κOO′:2κO′′)(formamide)2]n (1), [{Zn2(μ-L1-1κO:2κO′)2(4,4′-bipyridine)2(H2O)}·2(DMF)·5(H2O)]n (2), [Cd(μ-L1-μ-1κO2O′:2κO′′2O′′′)(DMF)]n (3), [{Zn(μ-L2-1κO:2κO′)(4,4′-bipyridine)(H2O)}·(H2O)]n (4) and [{Cd(μ-L2-μ-1κO2O′:2κO′′2O′′′)(formamide)(4,4′-bipyridine)}·(formamide)]n (5) [L1 = 5-propionamidoisophthalate, L2 = 5-benzamidoisophthalate, DMF = N,N-dimethylformamide]. These compounds have been characterized by elemental analysis, IR spectroscopy, thermogravimetric analysis and single-crystal X-ray diffraction. They act as heterogeneous catalysts in the transesterification reactions of various aromatic esters.
Results and discussion
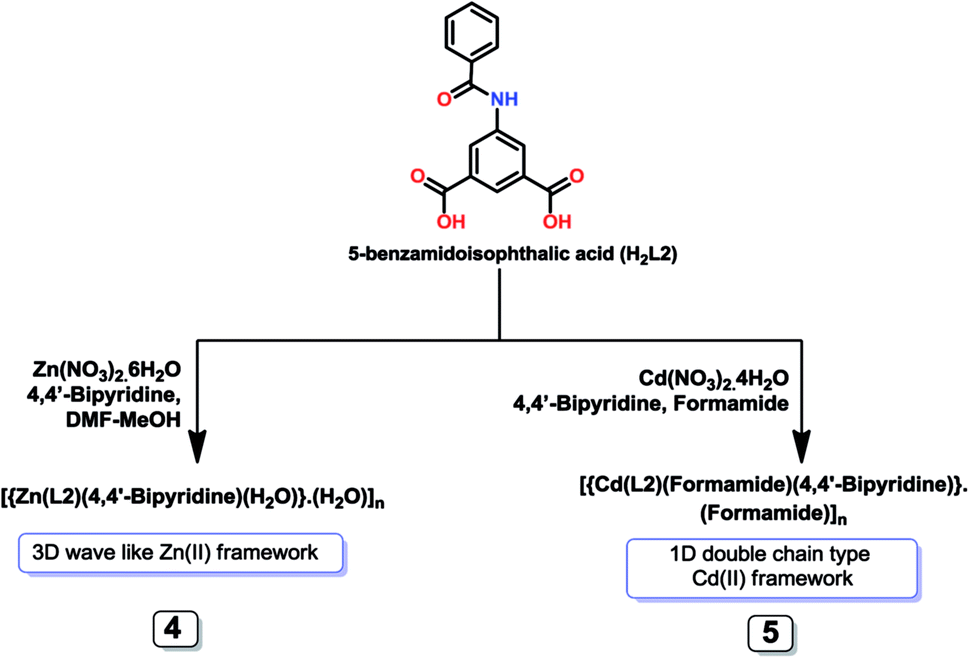
Syntheses and characterization
In general we have used solvothermal reaction procedures for the syntheses of these frameworks (1–5). The solvothermal reaction of H2L1 with zinc(II) nitrate hexahydrate in the presence of formamide lead to the formation of [Zn(μ-L1-μ-1κOO′:2κO′′)(formamide)2]n (1), but when in presence of 4,4′-bipyridine as auxiliary ligand and in a dimethylformamide (DMF) and methanol mixture, the two dimensional framework [{Zn2(μ-L1-1κO:2κO′)2(4,4′-bipyridine)2(H2O)}·2(DMF)·5(H2O)]n (2) is formed. The solvothermal reaction of Cd(NO3)2·4H2O with H2L1 in DMF, methanol and water mixture produced a 1D framework [Cd(μ-L1-μ-1κO2O′:2κO′′2O′′′)(DMF)]n (3) (Scheme 1). The solvothermal reaction of H2L2 with zinc(II) nitrate hexahydrate and 4,4′-bipyridine led to [{Zn(μ-L2-1κO:2κO′)(4,4′-bipyridine)(H2O)}·(H2O)]n (4), while the hydrothermal reaction of H2L2 and cadmium(II) nitrate in the presence of 4,4′-bipyridine as auxiliary ligand and in formamide, led to the formation of the one dimensional metal–organic framework [{Cd(μ-L2-μ-1κO2O′:2κO′′2O′′′)(formamide)(4,4′-bipyridine)}·(formamide)]n (5) (Scheme 2).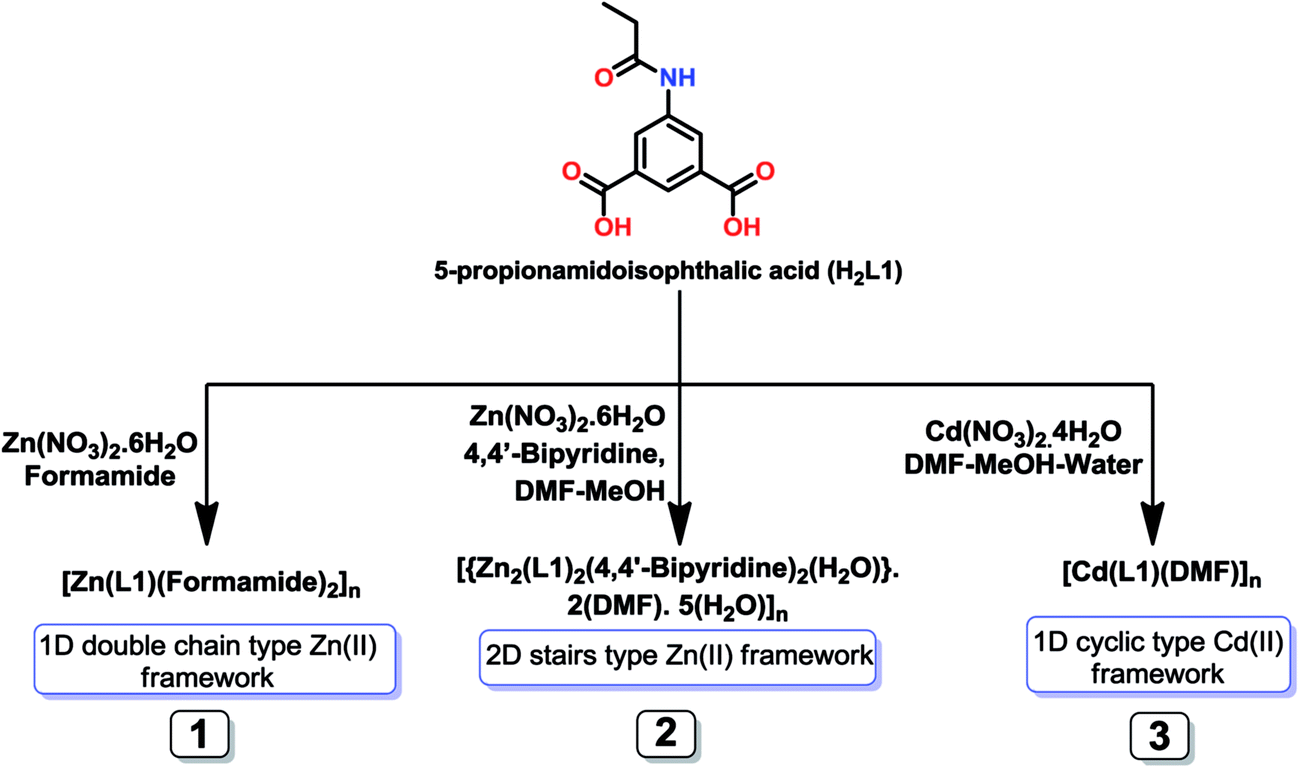 | ||
| Scheme 1 | ||
 | ||
| Scheme 2 | ||
In the IR spectra of 1–5, the characteristic strong bands of coordinated carboxylate groups appear at 1585–1560 cm−1 or 1388–1345 cm−1 for the asymmetric or the symmetric stretching, respectively.17 The bands in the regions 1635–1613 cm−1 and 1438–1418 cm−1 are attributed to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching frequencies of the aromatic rings, and those in the range 1689–1653 cm−1 to the stretching frequency of the amide CO group. Due to insolubility in common NMR solvents, these frameworks were only characterized by infrared, microanalysis and single crystal X-ray diffraction.
C stretching frequencies of the aromatic rings, and those in the range 1689–1653 cm−1 to the stretching frequency of the amide CO group. Due to insolubility in common NMR solvents, these frameworks were only characterized by infrared, microanalysis and single crystal X-ray diffraction.
Crystal structures analyses
According to the X-ray diffraction analysis, frameworks 1 and 5 have one dimensional double chain type structures, whereas framework 3 have 1D cyclic type structure, and 2 and 4 have very interesting two dimensional and 3D wave like structures, respectively.The asymmetric unit of 1 contains one deprotonated ligand (L12−), one Zn(II) cation and two coordinated formamide molecules (Fig. 1A and B). The metal cation has all-O trigonal bipyramidal geometry (τ5 = 0.66),18a with a basal plane made the O-atoms from both nonbridging monodentate and bridging bidentate carboxylate groups. While the former binding mode of the carboxylate groups leads to sixteen-membered C10O4Zn2 metallacycles, the latter assembles into C2O4Zn2 cores, in a chair conformation, which act as secondary building block units in the construction of the one dimensional double chain polymeric assembly (Fig. 1C). In this core, the Zn–OL1 assume values of 1.9882(17) and 1.9758(17) Å [Zn1–O1 and Zn1–O2, respectively], the coordination sphere of the metal being then fulfilled by the sole coordination of the other O-carboxylate group [Zn1–O4, 1.9713(18) Å] and by the O-atoms of formamide [Zn1–O6, 2.084(2) Å; Zn1–O7, 2.231(2) Å] molecules.
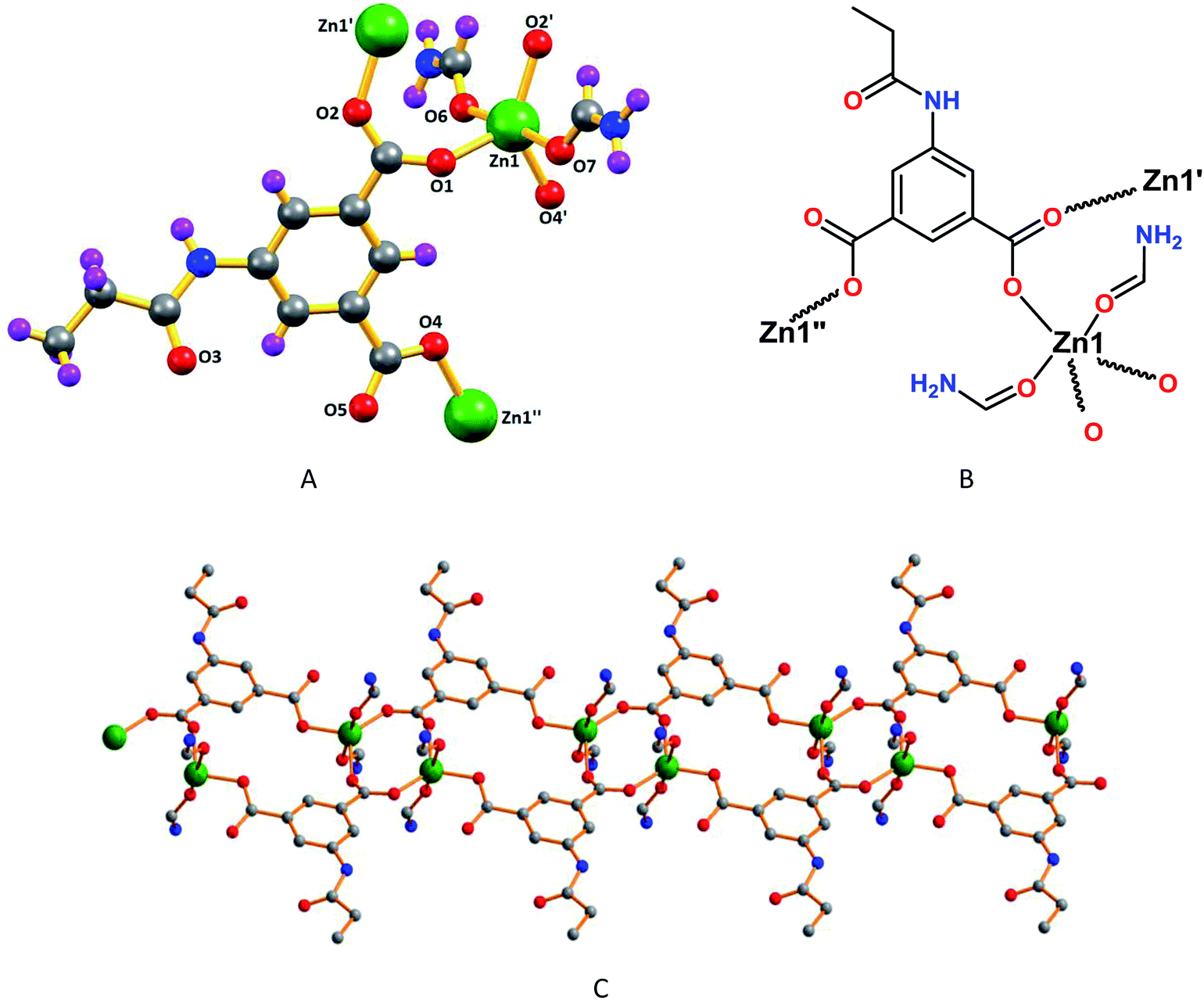 | ||
| Fig. 1 (A) Perspective view of framework 1 with partial atom labelling scheme and (B) its schematic representation. (C) One dimensional structure of 1. | ||
Framework 2 contains two Zn2+ ions bound to two deprotonated ligands (L12−), two 4,4′-bipyridine and one water molecule, the asymmetric unit of this compound also comprising two non-coordinated dimethylformamide and five water molecules (Fig. 2A).
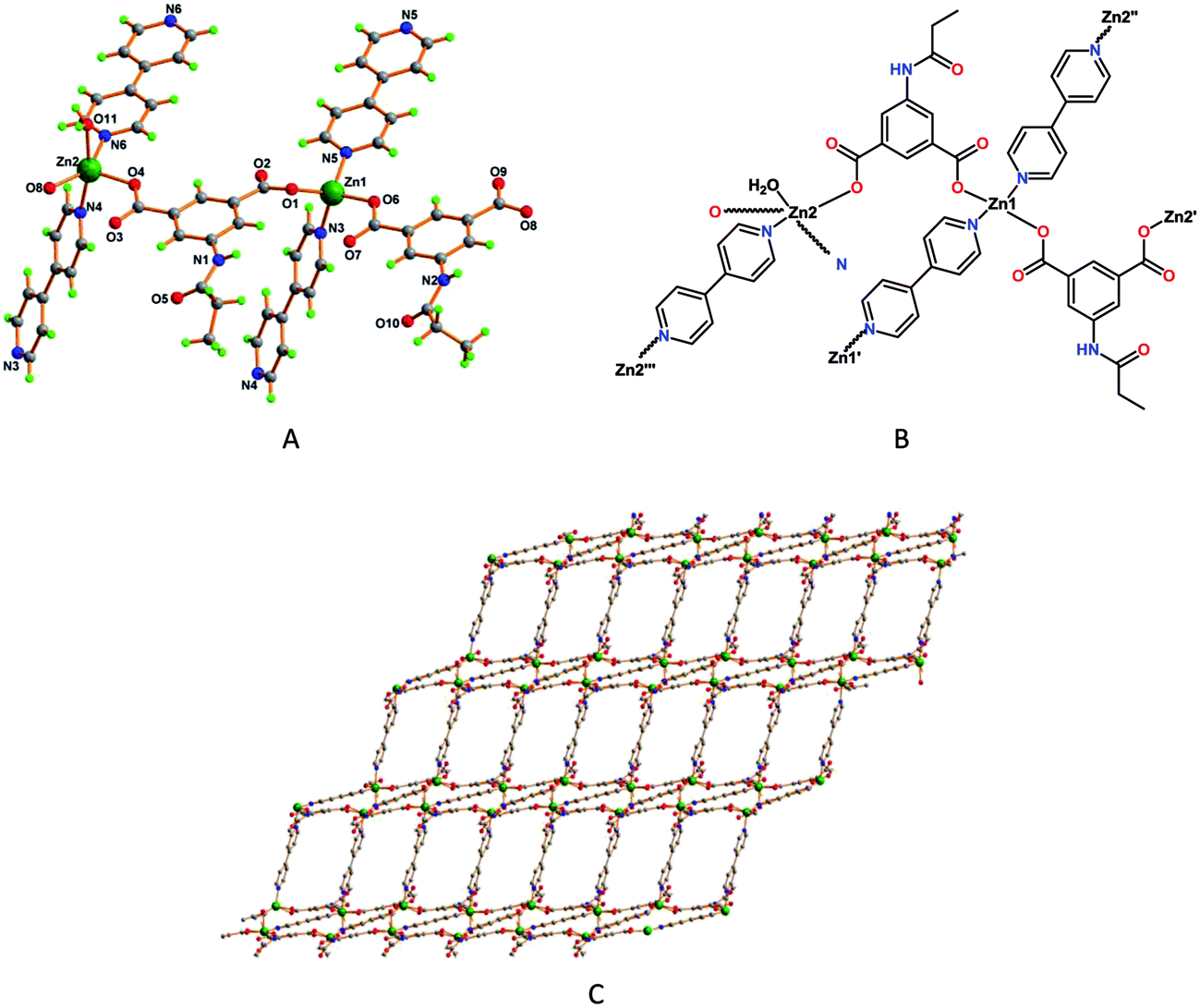 | ||
| Fig. 2 (A) Perspective view of framework 2 with partial atom labelling scheme, and (B) its schematic representation. (C) Two dimensional structure of framework 2. | ||
Framework 2 contains two different types of Zn(II) atoms with respect to their coordination environments [tetrahedral (τ4 = 0.88)18b for Zn1 and trigonal bipyramidal (τ5 = 0.38)18a for Zn2]. The Zn1 cation is coordinated to two of the O-atoms of two distinct L12− ligands [Zn1–O1, 1.9446(14) Å and Zn1–O6, 1.9373(13) Å] and further coordinated by the two N-atom of a bridging bipyridine moieties [Zn1–N3, 2.0539(17) Å and Zn1–N5, 2.0264(15) Å], which fulfill the tetrahedral coordination environment around the metal center (τ4 = 0.88).18b The coordination sphere around the Zn2 center fulfill by the coordination of two μ2-L12− ligands [Zn2–O4, 1.9871(13) Å and Zn2–O8, 1.9925(14) Å], two N-atom of a bridging bipyridine [Zn2–N4, 2.2311(17) Å and Zn2–N6, 2.0970(15) Å] and the remaining site fulfill by the water molecule [Zn2–O11, 2.2007(15) Å] (Fig. 2B). The carboxylate groups in 2 bind the metal in just the nonbridging monodentate fashion. Framework 2 features a stair type two-dimensional coordination polymeric chain, two L12− ligands bind two the two different metal atoms and forming a 1D chains which are further connected by two 4,4′-bipyridine molecules leading to the formation of 2D network (Fig. 2C), giving rise to tetranuclear C22N4O4Zn4 metalacycles.
The Cd2+ metal cations in 3 present all-O distorted seven coordinate pentagonal bipyramidal environments (Fig. 3A and B), each one binding four L1 units by means of six O-atoms from carboxylate groups working in bridging chelating fashions [Cd1–O3, 2.195(4) Å; Cd1–O4, 2.270(5) Å; Cd1–O1, 2.277(4) Å; Cd1–O2, 2.346(4) Å; Cd1–O1′, 2.414(4) Å]. The remaining coordination site is engaged with a formamide molecule [Cd1–O6, 2.3730(4) Å]. Compound 3 features a one-dimensional coordination polymeric chain with open channels, with approximate dimension of 2.3 × 2.3 Å2 (Fig. 3C), running along the crystallographic c-axis (Fig. 3D). The L1 ligand binds the metal in such a way that the flexible (amide) part, and the formamide moiety, is directed outwards such channels (Fig. 3D).
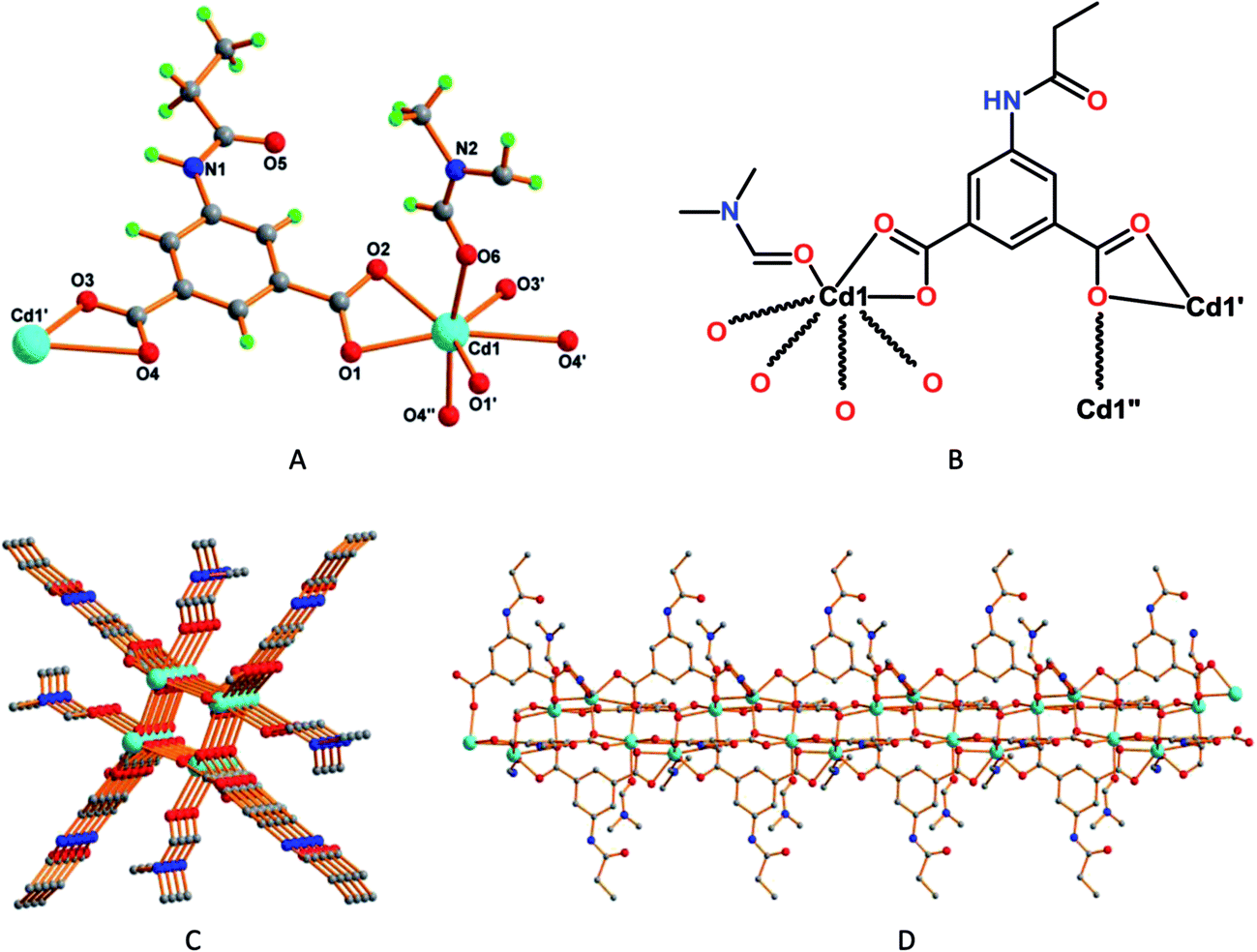 | ||
| Fig. 3 (A) Perspective view of framework 3 with partial atom labelling scheme, and (B) schematic representation of 3. (C) View of the one dimensional framework of 3 (arbitrary view) and (D) of the open channels running along the crystallographic c-axis. | ||
Single-crystal X-ray diffraction studies revealed that 4 is a 3D network with an asymmetric unit made of one Zn2+ ion bound to two L22− ligands, two halves of 4,4′-bipyridine and one water molecule, together with one non-coordinated water molecule (Fig. 4A). The Zn1 has trigonal bipyramidal coordination geometry (τ5 = 0.53),18a with the equatorial plane including two O atoms from two nonbridging monodentate carboxylate groups from two L22− ligands [Zn1–O3, 1.934(7) Å; Zn1–O6, 1.936(5) Å] and the N-atom from a 4,4′-bipyridine molecule [Zn1–N2, 2.051(7) Å]; the axial sites are occupied by the water molecule [Zn1–O1, 2.206(7) Å] and the N-atom of another 4,4′-bipyridine ligand [Zn1–N1, 2.234(6) Å] (Fig. 4B). In this framework one of the bipyridine molecules is almost planar, as measured by the nil angle between its two phenyl rings, but in the other that angle is of 38.03°. In addition, the phenyl groups of the L2 ligand make an angle of 73.92°. The bipyridine molecules and the 5-benzamidoisophthalate ligands are coordinated to the metal centers in such a way that a three dimensional wave like coordination networks is formed (Fig. 4C).
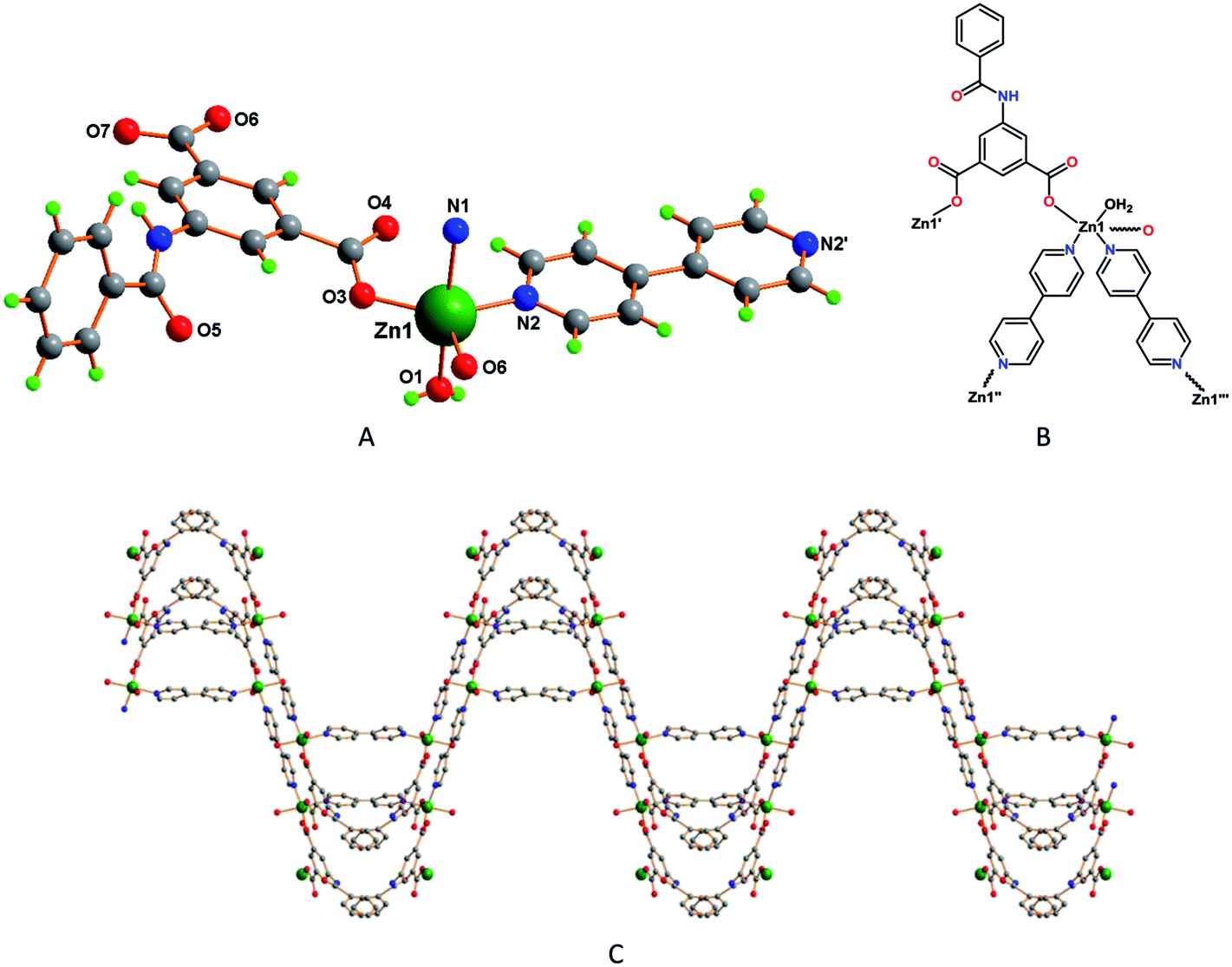 | ||
| Fig. 4 (A) Perspective view of framework 4 with partial atom labelling scheme (only one component of the disordered benzamido group is shown), and (B) its schematic representation. (C) Wave like three dimensional organisation of 4. | ||
When we perform the reaction of 5-benzamidoisophthalic acid (H2L2) with cadmium(II) nitrate in the presence of 4,4′-bipyridine in formamide, we have obtained another interesting one dimensional Cd(II) compound, framework 5. The asymmetric unit of 5 contains one Cd2+ ion coordinated to one deprotonated L2 ligand, one 4,4′-bipyridine and one formamide molecule, an additional non-coordinated formamide molecule also being present in the structure. The Cd1 center presents a distorted seven coordinated pentagonal bipyramidal environment (Fig. 5A) and binds to five carboxylate oxygen atoms of three neighboring L2 units, two of them in a bridging chelate [Cd1–O1 2.251(2) Å, Cd1–O1′ 2.551(2) Å, Cd1–O2 2.649(2) Å] and the remaining one in a chelate bidentate fashion [Cd1–O5 2.352(2) Å, Cd1–O6 2.3711(19) Å], and to one 4,4′-bipyridine molecule [Cd1–N2 2.265(2) Å] (Fig. 5B). In 5 the L2 ligand is almost planar and the phenyl rings of 4,4′-bipyridine are relatively twisted by 39.82°, features that conceivably prevented the formation of a structure with a higher dimensionality (as in 4) and a 1D double chain type structure is formed instead (Fig. 5C).
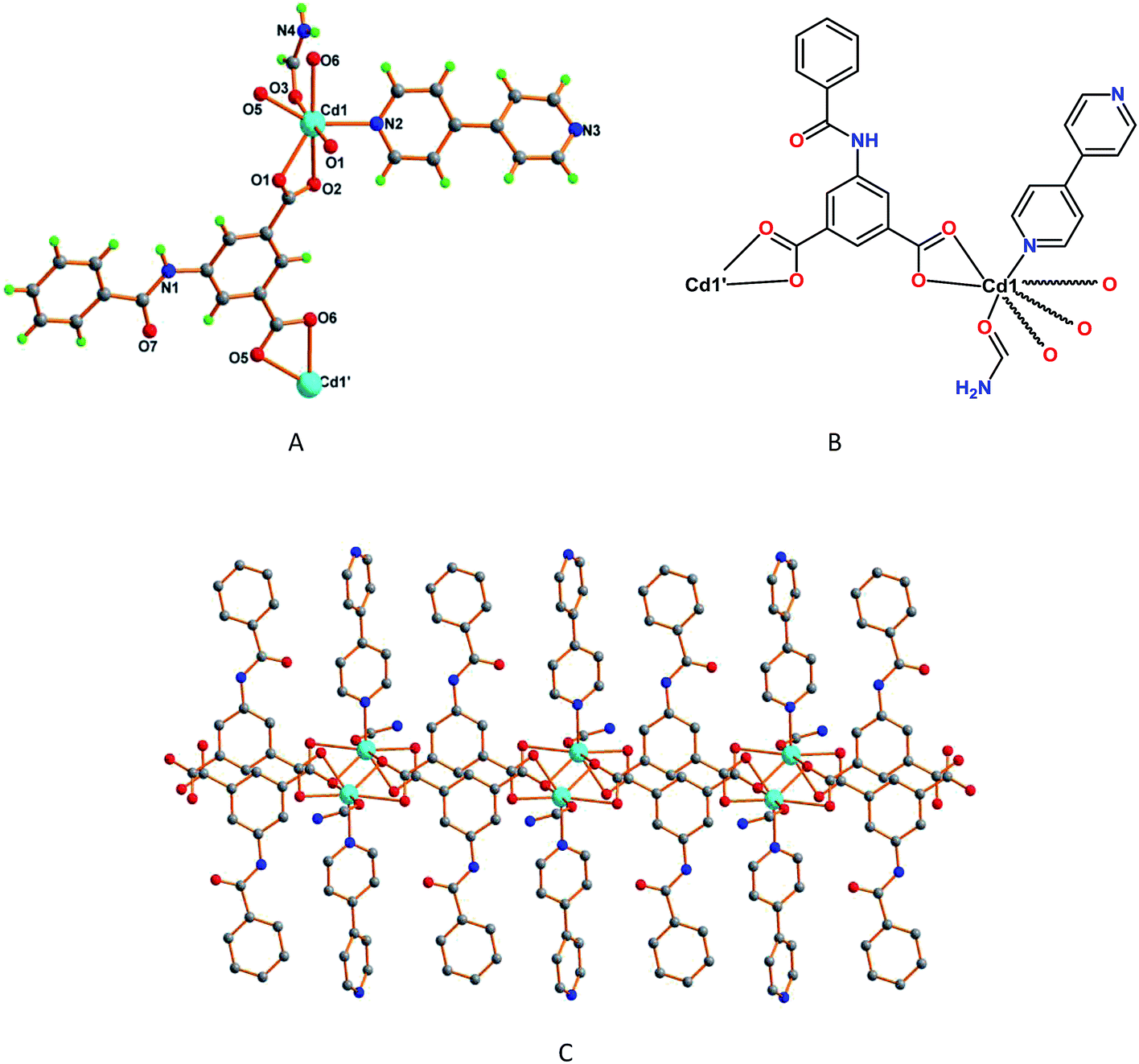 | ||
| Fig. 5 (A) A view of framework 5 with partial atom labelling scheme, and (B) schematic representation of 5. (C) One dimensional double chain type structure of 5. | ||
To improve the description of the crystal structures of 1–5 we performed their topological analysis by reducing their multidimensional structures to simple node-and-linker nets where the metallic nodes and the organic linkers represent secondary building units (SBUs).19 We have carried out these analyses by using TOPOS 4.0.19b Frameworks 1 and 5 reveals that they have 3-connected uninodal nets with topological SP1-periodic types (Fig. 6A), 2 has a 2,4-connected binodal (Fig. 6B) and 3 a 4-connected uninodal net (Fig. 6D), but 4 has a more complex 2,2,2,4-connected tetranodal structure (Fig. 6C).
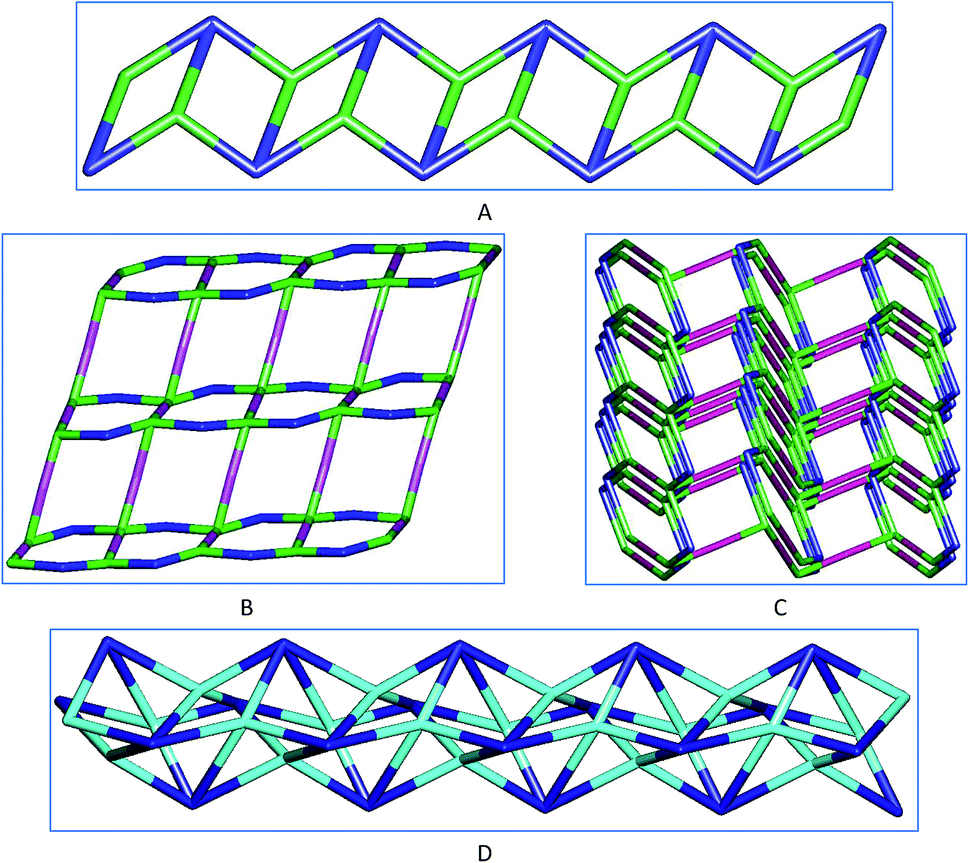 | ||
| Fig. 6 Node-and-linker-type descriptions of the 1D coordination frameworks in 1 and 5 (A), 2D in 2 (B), 3D in 4 (C) and 1D in 3 (D). The metal nodes are represented in green (Zn) and cyano (Cd) color, carboxylate linker in blue color and the bipyridine in pink color. | ||
Thermogravimetric analyses
Thermogravimetric analyses were carried out under dinitrogen in the range from room temperature to ca. 750 °C at a heating rate of 5 °C min−1. Features of the thermal stability of complex 1–5 are illustrated in Fig. 7.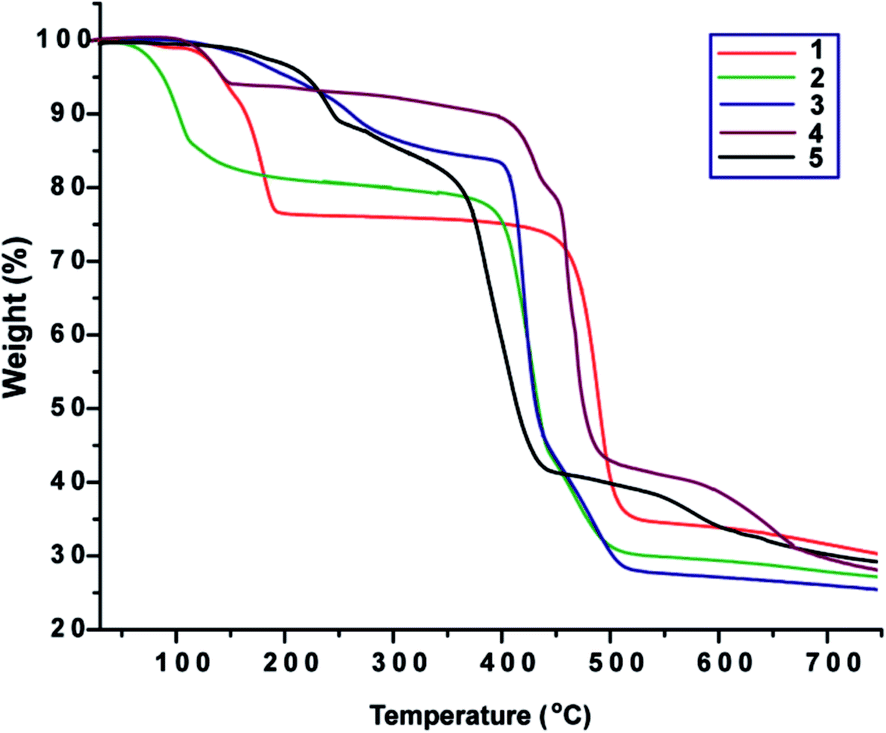 | ||
| Fig. 7 Thermogravimetric curves for 1–5. | ||
Compound 1 shows two step decompositions and loses 23.7% of its weight within 102–204 °C, due to loss of two coordinated formamide molecules (calcd: 23.0%). The remaining material then started to decompose from 409 °C until 538 °C. Compound 2 shows a weight loss of 21.5% within 41–248 °C, corresponding to the loss of six molecules of water and two dimethylformamide molecules (calcd: 21.7%). Upon further heating, it is stable up to 362 °C and then started to decompose. Compound 3 exhibits a weight loss of 17.1% in the 99–333 °C temperature range, which accounts for the total removal of one dimethylformamide molecule (calcd: 17.3%). The remaining material then started to decompose from 380 °C until 523 °C. Compound 4 shows a weight loss of 7.1% in the 97–158 °C range, corresponding to the loss of two molecules of water (calcd: 6.6%). Compound 5 shows a continuous decomposition and loses 15.1% of its weight within 153–298 °C, most likely due to loss of two formamide molecules (calcd: 14.0%). The remaining material continues decomposing until 468 °C.
Transesterification
Due to the very low solubility of 1–5 in alcohols, they are potentially good candidates for heterogeneous catalytic transesterification reactions. We have tested their catalytic activities for this reaction by using a mixture of methyl-4-nitrobenzoate in 2 mL EtOH with the catalyst (1–5) contained in a capped glass vessel and stirred at 75 °C, typically for 30 h (Scheme 3). The catalyst was then removed by centrifugation and the solvent was evaporated in vacuum, leading to a crude mixture of products which was analyzed by 1H NMR (Table 1).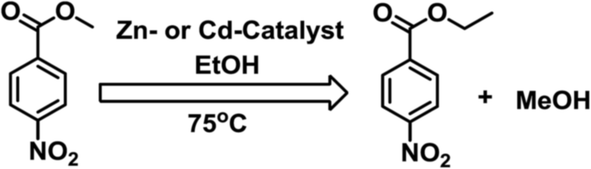 | ||
| Scheme 3 Transesterification reaction. | ||
| Entry | Catalyst | Time (h) | Amount of catalystb (mol%) | T (°C) | Alcohol | Yieldc (%) |
|---|---|---|---|---|---|---|
| a Reaction conditions, unless stated otherwise: 2.0 mol% of catalyst, methyl-4-nitrobenzoate (1.0 mmol) and alcohol (2.0 mL) at 75 °C for 30 h.b Relative to substrate.c Calculated by 1H NMR. | ||||||
| 1 | 4 | 1 | 2.0 | 75 | EtOH | 12 |
| 2 | 4 | 2 | 2.0 | 75 | EtOH | 21 |
| 3 | 4 | 4 | 2.0 | 75 | EtOH | 48 |
| 4 | 4 | 8 | 2.0 | 75 | EtOH | 72 |
| 5 | 4 | 12 | 2.0 | 75 | EtOH | 85 |
| 6 | 4 | 30 | 2.0 | 75 | EtOH | 91 |
| 7 | 4 | 48 | 2.0 | 75 | EtOH | 92 |
| 8 | 4 | 30 | 1.0 | 75 | EtOH | 65 |
| 9 | 4 | 30 | 5.0 | 75 | EtOH | 92 |
| 10 | 4 | 30 | 2.0 | RT | EtOH | 5 |
| 11 | 4 | 30 | 2.0 | 50 | EtOH | 21 |
| 12 | 4 | 30 | 2.0 | 100 | EtOH | 89 |
| 13 | 4 | 30 | 2.0 | 75 | 1-PrOH | 56 |
| 14 | 4 | 30 | 2.0 | 75 | 2-PrOH | 38 |
| 15 | 1 | 30 | 2.0 | 75 | EtOH | 78 |
| 16 | 2 | 30 | 2.0 | 75 | EtOH | 81 |
| 17 | 3 | 30 | 2.0 | 75 | EtOH | 65 |
| 18 | 5 | 30 | 2.0 | 75 | EtOH | 54 |
| 19 | Blank | 30 | — | 75 | EtOH | — |
| 20 | Zn(NO3)2·6H2O | 30 | 2.0 | 75 | EtOH | 11 |
| 21 | Cd(NO3)2·4H2O | 30 | 2.0 | 75 | EtOH | 7 |
| 22 | H2L1 | 30 | 2.0 | 75 | EtOH | — |
| 23 | H2L2 | 30 | 2.0 | 75 | EtOH | — |
We have optimized the transesterification reaction conditions (temperature, reaction time, amount of catalyst and solvent) using 4 as the catalyst since we found that this framework showed a higher activity than the others (1, 2, 3 and 5) after the same reaction time and at the same temperature.
The increase of the catalyst amount from 1.0 and 2.0 mol% improves the product yield from 65% to 91% (30 h reaction time), but a further increase in the catalyst amount to 5 mol% did not increase the yield significantly (entries 6, 8, 9, Table 1). The highest yield was achieved with ethanol (91% in 30 h, entry 6, Table 1), while yields of 56% and 38% were obtained with 1-propanol and 2-propanol, respectively (entries 13 and 14, Table 1). Increasing the reaction temperature from room temperature to 75 °C increased the yield, e.g., from 5% to 91% for ethanol but a further rise in the temperature led to a slight yield decrease (entries 6 and 10–12, Table 1). The product yield vs. time plot is shown in Fig. 8A.
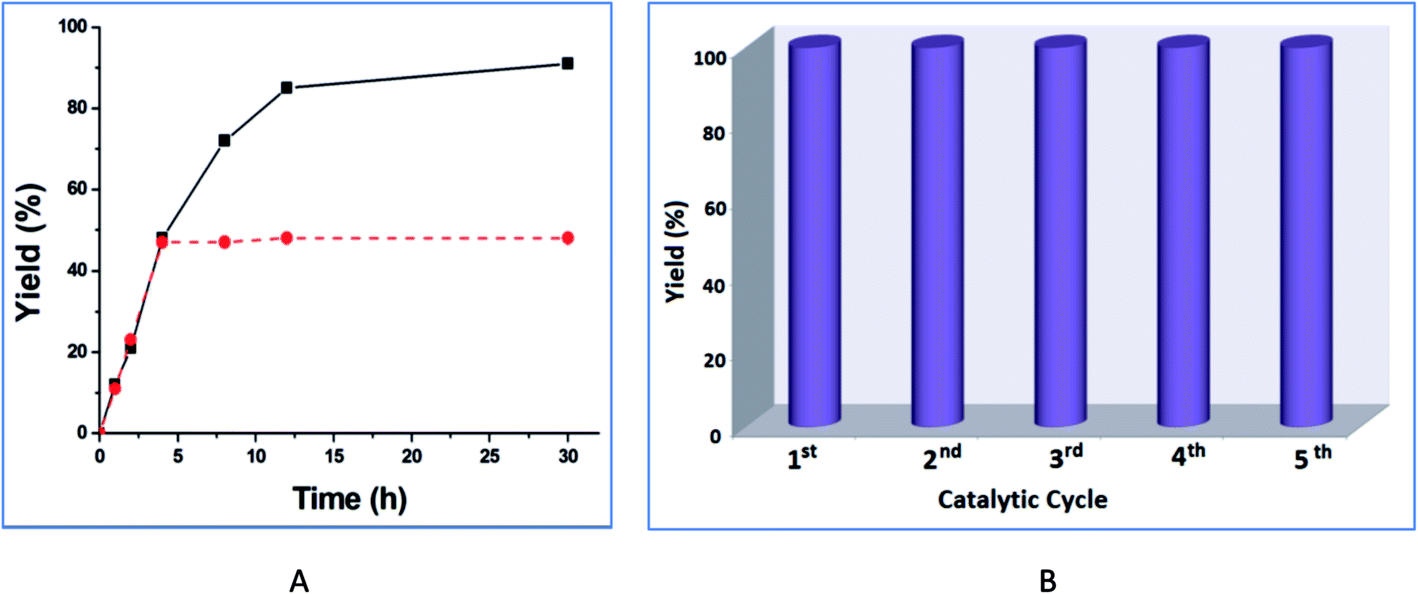 | ||
| Fig. 8 (A) Plot of product yield vs. time for the transesterification reaction of methyl-4-nitrobenzoate and ethanol with 4 at 75 °C (dotted line: removal of catalyst after 4 h, see text). (B) Effect of the catalyst recycling on the yield of ethyl-4-nitrobenzoate. | ||
The reaction between methyl-4-nitrobenzoate and ethanol was monitored in blank, also using the free ligand precursors H2L1 and H2L2 as well as the metal salts Zn(NO3)2·6H2O and Cd(NO3)2·4H2O, instead of the catalysts (entries 19–23, Table 1). The reaction did not progress at all in the absence of any metal compound, and only 11% or 7% of product yield was obtained when using Zn(NO3)2·6H2O or Cd(NO3)2·4H2O as catalyst, respectively.
Under the optimized reaction conditions, when 2 mol% of compound 4 is used as catalyst, a conversion of 91% of methyl-4-nitrobenzoate into ethyl-4-nitrobenzoate is reached after 30 h at 75 °C (entry 6, Table 1). Lengthening the reaction time to 48 h did not improve the yield of the reaction significantly. Under these optimized conditions but using frameworks 1, 2, 3 and 5 as catalysts, conversions of 78%, 81%, 65% and 54% (entries 15–18, Table 1) were obtained, respectively. Catalyst 4 is the most effective one, the catalytic performance following the order of 4 > 2 > 1 > 3 > 5 under the same experimental conditions (Table 1, entries 7, 15–18). Hence the Zn-MOFs are better catalysts than the Cd ones. Although a relationship between the catalysts structure and their activity towards the transesterification reaction cannot be ascertained, it is clear that the Zn-frameworks 1, 2 and 4 have better performances as compared to the Cd 3 and 5, and among the former, compound 4 is the best. Apart from the central metal cation, the three dimensional packing structure in 4 may also play a role.
Once having established that compound 4 is the best catalyst for the transesterification of methyl-4-nitrobenzoate, we investigated the reaction of other methyl benzoates in ethanol (Table 2). The substrates with the strongest electron-withdrawing nitro substituent show the highest activity (entry 2, Table 2 and entry 6, Table 1) while that with the electron-donating methoxy group exhibits the lowest one (entry 6, Table 2).
| Entry | Substrate | Yieldb (%) |
|---|---|---|
| a Reaction conditions: 2.0 mol% of catalyst 4, methyl ester (1.0 mmol) and ethanol (2.0 mL) at 75 °C for 30 h.b Calculated by 1H NMR. | ||
| 1 | Methyl benzoate | 68 |
| 2 | Methyl-3-nitrobenzoate | 89 |
| 3 | Methyl-4-aminobenzoate | 71 |
| 4 | Methyl-3-hydroxybenzoate | 65 |
| 5 | Methyl-4-hydroxybenzoate | 62 |
| 6 | Methyl-4-methoxybenzoate | 52 |
Leaching in general is a common phenomenon observed in the heterogeneous catalytic systems. In order to test the possibility of leaching and the heterogeneity of our catalytic system, an experiment was performed following the procedure described by Sheldon et al.20 After the reaction mixture was stirred for 4 h at 75 °C (only ca. 48% conversion of methyl-4-nitrobenzoate was then observed), the catalyst was removed by filtration and no further conversion was observed (Fig. 8A). These results support the heterogeneity of the catalyst with no appreciable leaching. Additionally, we have determined the amount of zinc in the filtrated solution after the separation of the catalyst 4. The result shows only 0.03% of zinc is present in the solution, thus ruling out any significant leaching of the catalyst.
In order to examine the recyclability of catalyst 4, we removed it by filtration after the transesterification reaction of methyl-4-nitrobenzoate in ethanol, dried it and reused it as catalyst in a subsequent transesterification process. The catalytic system maintained constant activity for five consecutive cycles (Fig. 8B). FT-IR and PXRD spectra of 4 taken before and after the reaction (ESI, Fig. S2†) did not indicate any important changes. This suggests that the integrity of catalyst 4 was retained after the reaction.
A possible mechanism for the transesterification catalyzed by 1–5 is presented in Scheme 4, on the basis of reported proposals.10a,16c Coordination of the benzoate ester R1COOMe to the Lewis acid metal centre leads to the increase of the electrophilicity of the carbonyl carbon which is thus activated towards nucleophilic attack by the alcohol R2OH.
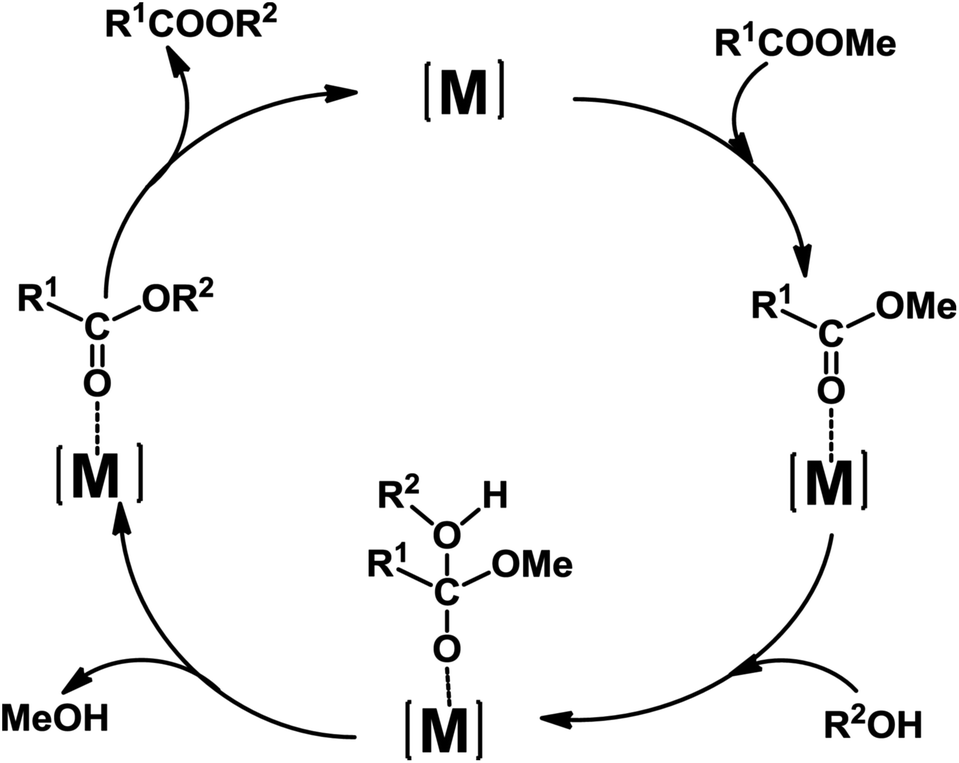 | ||
| Scheme 4 Proposed catalytic cycle for the transesterification reaction of methyl benzoates catalyzed by 1–5 ([M] = Zn(II) or Cd(II) centre; R1 = benzoyl or substituted benzoyl; R2 = Et or Pr). | ||
Conclusion
We successfully synthesized five different metal–organic frameworks (1–5) of zinc(II) or cadmium(II) from 5-propionamidoisophthalic acid (H2L1) or 5-benzamidoisophthalic acid (H2L2) in presence or absence of an auxiliary ligand under various synthetic conditions. They were structurally characterized by single crystal X-ray diffraction analyses which reveal that they exhibit 1D, 2D or 3D structures with interesting topologies. Compounds 1 and 5 possess one dimensional double chain type structures, whereas framework 3 has a 1D cyclic type structure. Compounds 2 and 4 have two dimensional and three dimensional wave like structures, respectively.These frameworks (1–5) act as good heterogeneous catalysts in the transesterification reactions of methyl carboxylate esters. The Zn-MOFs display a higher catalytic activity than the Cd ones and 4, with a 3D wave like structure, is the most active one. It effectively catalyzes the reaction of various methyl benzoate esters with ethanol producing the corresponding ethyl benzoate esters (and methanol) in high yields which depend on the electrophilicity of the benzoate substituent. The stability and recyclability of the heterogeneous catalysts were proved. Hence, MOFs containing Zn(II) ions can be utilized as effective heterogeneous catalysts for the transesterification reaction, with “green” features that deserve to be further explored for other reactions.
Experimental
All the chemicals were obtained from commercial sources and used as received. The infrared spectra (4000–400 cm−1) were recorded on a Bruker Vertex 70 instrument in KBr pellets; abbreviations: s = strong, m = medium, w = weak, bs = broad and strong, mb = medium and broad. Carbon, hydrogen, and nitrogen elemental analyses were carried out by the Microanalytical Service of the Instituto Superior Técnico. Thermogravimetric analyses were carried out with a Perkin-Elmer Instrument system (STA6000) at a heating rate of 10 °C min−1 under a dinitrogen atmosphere, in the range of room temperature up to ca. 750 °C. Powder X-ray diffraction (PXRD) was conducted in a D8 Advance Bruker AXS (Bragg–Brentano geometry) theta–2theta diffractometer, with copper radiation (Cu Kα, λ = 1.5406 Å) and a secondary monochromator, operated at 40 kV and 40 mA. Flat plate configuration was used and the typical data collection range was between 5° and 40°. The synthetic work was performed in air and at elevated temperature.Synthesis of 1
A mixture of Zn(NO3)2·6H2O (25.0 mg, 0.084 mmol) and H2L1 (20.0 mg, 0.084 mmol) was dissolved in 1 mL of formamide. The resulting mixture was sealed in an 8 mL glass vessel and heated at 75 °C for 48 h. Subsequent gradual cooling to room temperature (0.2 °C min−1) afforded colorless crystals. Yield: 73% (based on Zn). Anal. calcd for C13H15N3O7Zn (M = 390.65): C, 39.97; H, 3.87; N, 10.76; found: C, 38.82; H, 3.65; N, 9.82. FT-IR (KBr, cm−1): 3393 (bs), 3320 (mb), 2986 (w), 2946 (w), 1674 (s), 1635 (s), 1578 (s), 1438 (s), 1388 (s), 1318 (m), 1220 (s), 1115 (w), 789 (s), 723 (s), 653 (w), 626 (s), 458 (m).Synthesis of 2
A mixture of Zn(NO3)2·6H2O (25.0 mg, 0.084 mmol), H2L1 (20.0 mg, 0.084 mmol) and 4,4′-bipyridine (13.1 mg, 0.084 mmol) was dissolved in 1 mL of DMF and methanol (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). A white precipitate was obtained when 0.5 mL of 28% aqueous ammonia solution was added to this reaction mixture. The precipitate was dissolved upon the addition of additional 0.5 mL of 28% aqueous ammonia solution. Then, the resulting mixture was sealed in an 8 mL glass vessel and heated at 75 °C for 48 h. It was subsequently cooled to room temperature (0.2 °C min−1), affording block-like colorless crystals. Yield: 61% (based on Zn). Anal. calcd for C48H60N8O18Zn2 (M = 1167.79): C, 49.37; H, 5.18; N, 9.60; found: C, 49.11; H, 5.01; N, 9.95. FT-IR (KBr, cm−1) 3409 (bs), 3279 (bs), 2970 (w), 2933 (w), 1689 (s), 1664 (s), 1613 (s), 1585 (s), 1492 (m), 1421 (s), 1345 (s), 1224 (w), 1103 (w), 1073 (m), 829 (w), 780 (s), 729 (s), 639 (m).
:1). A white precipitate was obtained when 0.5 mL of 28% aqueous ammonia solution was added to this reaction mixture. The precipitate was dissolved upon the addition of additional 0.5 mL of 28% aqueous ammonia solution. Then, the resulting mixture was sealed in an 8 mL glass vessel and heated at 75 °C for 48 h. It was subsequently cooled to room temperature (0.2 °C min−1), affording block-like colorless crystals. Yield: 61% (based on Zn). Anal. calcd for C48H60N8O18Zn2 (M = 1167.79): C, 49.37; H, 5.18; N, 9.60; found: C, 49.11; H, 5.01; N, 9.95. FT-IR (KBr, cm−1) 3409 (bs), 3279 (bs), 2970 (w), 2933 (w), 1689 (s), 1664 (s), 1613 (s), 1585 (s), 1492 (m), 1421 (s), 1345 (s), 1224 (w), 1103 (w), 1073 (m), 829 (w), 780 (s), 729 (s), 639 (m).
Synthesis of 3
A mixture of Cd(NO3)2·4H2O (25.9 mg, 0.084 mmol) and H2L1 (20 mg, 0.084 mmol) was dissolved in 2 mL of DMF, methanol and water (0.5:1:0.5) mixture. The resulting mixture was sealed in an 8 mL glass vessel and heated at 75 °C for 48 h. It was subsequently cooled to room temperature (0.2 °C min−1), affording colorless crystals. Yield: 68% (based on Cd). Anal. calcd for C14H16CdN2O6 (M = 420.70): C, 39.97; H, 3.83; N, 6.66; found: C, 39.88; H, 3.11; N, 6.10. FT-IR (KBr, cm−1): 3323 (bs), 3083 (b), 2983 (w), 2929 (w), 1659 (s), 1616 (s), 1567 (s), 1435 (s), 1366 (s), 1322 (m), 1244 (s), 1103 (s), 920 (w), 811 (m), 783 (s), 730 (s), 671 (w), 456 (m).
Synthesis of 4
A mixture of Zn(NO3)2·6H2O (20.7 mg, 0.07 mmol), H2L2 (20.0 mg, 0.07 mmol) and 4,4′-bipyridine (11.1 mg, 0.07 mmol) was dissolved in 2 mL of DMF and methanol (1:1). A white precipitate was obtained when the mixture was stirred at room temperature. The precipitate was dissolved upon the addition of 0.5 mL of 28% aqueous ammonia solution. Then, the resulting mixture was sealed in an 8 mL glass vessel and heated at 75 °C for 48 h. It was subsequently cooled to room temperature (0.2 °C min−1), affording block-like colorless crystals. Yield: 49% (based on Zn). Anal. calcd for C25H21N3O7Zn (M = 540.83): C, 55.52; H, 3.91; N, 7.77; found: C, 55.21; H, 3.92; N, 7.10. FT-IR (KBr, cm−1): 3404 (bs), 326 (bs), 1683 (s), 1617 (s), 1562 (s), 1454 (m), 1418 (s), 1382 (s), 1232 (m), 1107 (w), 1054 (w), 771 (s), 732 (s), 627 (m), 460 (w).
Synthesis of 5
A mixture of Cd(NO3)2·4H2O (31.0 mg, 0.10 mmol), H2L1 (28.5 mg, 0.10 mmol) and 4,4′-bipyridine (15.6 mg, 0.10 mmol) was dissolved in 2 mL of formamide. Then, the resulting mixture was sealed in an 8 mL glass vessel and heated at 75 °C for 48 h. It was subsequently cooled to room temperature (0.2 °C min−1), affording block-like colorless crystals. Yield: 79% (based on Cd). Anal. calcd for C27H23CdN5O7 (M = 641.91): C, 50.52; H, 3.61; N, 10.91; found: C, 50.83; H, 3.92; N, 10.09. FT-IR (KBr, cm−1): 3392 (bs), 3281 (bs), 1653 (s), 1560 (s), 1491 (m), 1429 (s), 1367 (s), 1285 (m), 1228 (m), 1105 (w), 1069 (w), 777 (s), 733 (s), 625 (m).Crystal structure determinations
X-ray quality single crystals of the compounds were immersed in cryo-oil, mounted in a nylon loop and measured at room temperature (1–5). Intensity data were collected using a Bruker APEX-II PHOTON 100 diffractometer with graphite monochromated Mo-Kα (λ 0.71069) radiation. Data were collected using phi and omega scans of 0.5° per frame and a full sphere of data was obtained. Cell parameters were retrieved using Bruker SMART21 software and refined using Bruker SAINT21a on all the observed reflections. Absorption corrections were applied using SADABS.21a Structures were solved by direct methods by using the SHELXS-97 package21b and refined with SHELXL-2014/7.21b Calculations were performed using the WinGX System-Version 1.80.03.21c The hydrogen atoms attached to carbon atoms were inserted at geometrically calculated positions and included in the refinement using the riding-model approximation; Uiso(H) were defined as 1.2Ueq of the parent nitrogen atoms or the carbon atoms for phenyl and methylene residues, and 1.5Ueq of the parent carbon atoms for the methyl groups. The hydrogen atoms of coordinated water molecules were located from the final difference Fourier map and the isotropic thermal parameters were set at 1.5 times the average thermal parameters of the belonging oxygen atoms and their distances were fixed using DFIX and DANG. The hydrogen atoms of the non-coordinated water were inserted in calculated positions by means of the CALC-OH routine of WinGX but their distances were fixed using DFIX and DANG to achieve a reasonable result. In compound 4, the banzamido group is highly disordered between two different positions: the phenyl group and the N-amide atom were modelled by means of PART 1 and PART 2 instructions leading to occupancies of 45.8 and 54.2% for the disordered components; the strategy for the O-amide atom was different, with the s.o.f. of both components set to 10.5000, one of them flanked by PART 1 and PART 0 to exclude bonds to symmetry related atoms. Least square refinements with anisotropic thermal motion parameters for all the non-hydrogen atoms and isotropic ones for the remaining atoms were employed. Crystallographic data are summarized in Table S1 (ESI file†) and selected bond distances and angles are presented in Table S2.† CCDC 1490728–1490732 contain the supplementary crystallographic data for this paper.†Procedure for the transesterification reaction catalyzed by MOFs
A mixture of methyl-4-nitrobenzoate (180 mg, 1 mmol) and MOF-catalyst (8 mg for 1, 23 mg of 2, 8 mg of 3, 11 mg of 4 and 13 mg of 5, 2 mol%) in 2 mL EtOH was added into a capped glass vessel. The mixture was heated at 75 °C for the desired time (typically 30 h). The solution was then centrifuged to remove the catalyst. The solvent was evaporated in vacuum, leading to a crude product. The product mixture was analyzed by 1H NMR in CDCl3. The yield of the ethyl ester product (relatively to the methyl ester) was established typically by taking into consideration the relative amounts of these compounds, as given by 1H NMR. The 1H-NMR spectra are presented in Fig. S1 (ESI file†). The centrifuged catalyst was washed several time with methanol and dried at room temperature. After that the recycling experiment was performed under the conditions mentioned above.Acknowledgements
This work has been supported by the Foundation for Science and Technology (FCT) (project UID/QUI/00100/2013), Portugal. Authors A. Karmakar and A. P. C. Ribeiro express his gratitude to the FCT for post-doctoral fellowship (Ref. No. SFRH/BPD/76192/2011 and SFRH/BPD/90883/2012).References
- (a) H.-C. Zhou and S. Kitagawa, Chem. Soc. Rev., 2014, 43, 5415–5418 RSCZ. Zhang and M. J. Zaworotko, Chem. Soc. Rev., 2014, 43, 5444–5455 RSC; (b) A. Karmakar, H. M. Titi and I. Goldberg, Cryst. Growth Des., 2011, 11, 2621–2636 CrossRef CAS; (c) M. P. Suh, H. J. Park, T. K. Prasad and D.-W. Lim, Chem. Rev., 2012, 112, 782–835 CrossRef CAS PubMed; (d) J. R. Long and O. M. Yaghi, Chem. Soc. Rev., 2009, 38, 1213–1214 RSC; (e) H.-C. Zhou, J. R. Long and O. M. Yaghi, Chem. Rev., 2012, 112, 673–674 CrossRef CAS PubMed; (f) Y. He, W. Zhou, G. Qian and B. Chen, Chem. Soc. Rev., 2014, 43, 5657–5678 RSC; (g) S. Furukawa, J. Reboul, S. Diring, K. Sumida and S. Kitagawa, Chem. Soc. Rev., 2014, 43, 5700–5734 RSC; (h) Q.-L. Zhu and Q. Xu, Chem. Soc. Rev., 2014, 43, 5468–5512 RSC.
- (a) O. R. Evans and W. Lin, Acc. Chem. Res., 2002, 35, 511–522 CrossRef CAS PubMed; (b) N. L. Rosi, J. Eckert, M. Eddaoudi, D. T. Vodak, J. Kim, M. O'Keeffe and O. M. Yaghi, Science, 2003, 300, 1127–1129 CrossRef CAS PubMed; (c) B. Kesanli, Y. Cui, M. R. Smith, E. W. Bittner, B. C. Bockrath and W. Lin, Angew. Chem., Int. Ed., 2005, 44, 72–75 CrossRef CAS PubMed; (d) C.-D. Wu and W. Lin, Angew. Chem., Int. Ed., 2007, 46, 1075–1078 CrossRef CAS PubMed; (e) D. N. Dybtsev, A. L. Nuzhdin, H. Chun, K. P. Bryliakov, E. P. Talsi, V. P. Fedin and K. Kim, Angew. Chem., Int. Ed., 2006, 45, 916–920 CrossRef CAS PubMed; (f) W. Lin, J. Solid State Chem., 2005, 178, 2486–2490 CrossRef CAS; (g) A. J. Fletcher, E. J. Cussen, D. Bradshaw, M. J. Rosseinsky and K. M. Thomas, J. Am. Chem. Soc., 2004, 126, 9750–9759 CrossRef CAS PubMed; (h) L. Alaerts, C. E. A. Kirschhock, M. Maes, M. A. van der Veen, V. Finsy, A. Depla, J. A. Martens, G. V. Baron, P. A. Jacobs, J. E. M. Denayer and D. E. De Vos, Angew. Chem., Int. Ed., 2007, 46, 4293–4297 CrossRef CAS PubMed.
- (a) A. Dhakshinamoorthy and H. Garcia, Chem. Soc. Rev., 2014, 43, 5750–5765 RSC; (b) L. Ma, C. Abney and W. Lin, Chem. Soc. Rev., 2009, 38, 1248–1256 RSC; (c) J. Y. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450–1459 RSC; (d) A. Karmakar, H. M. Titi and I. Goldberg, Cryst. Growth Des., 2011, 11, 2621–2636 CrossRef CAS; (e) A. Dhakshinamoorthy, M. Opanasenko, J. Cejka and H. Garcia, Adv. Synth. Catal., 2013, 355, 247–268 CAS.
- (a) H. L. Jiang, Y. Tatsu, Z. H. Lu and Q. Xu, J. Am. Chem. Soc., 2010, 132, 5586–5587 CrossRef CAS PubMed; (b) M. H. Zeng, W. X. Zhang, X. Z. Sun and X. M. Chen, Angew. Chem., Int. Ed., 2005, 44, 3079–3082 CrossRef CAS PubMed; (c) P. J. Steel, Acc. Chem. Res., 2005, 38, 243–250 CrossRef CAS PubMed; (d) P. M. Forster, N. Stock and A. K. Cheetham, Angew. Chem., Int. Ed., 2005, 44, 7608–7611 CrossRef CAS PubMed; (e) G. C. Ou, X. L. Feng and T. B. Lu, Cryst. Growth Des., 2011, 11, 851–856 CrossRef CAS; (f) J. Ni, K. J. Wei, Y. Liu, X. C. Huang and D. Li, Cryst. Growth Des., 2010, 10, 3964–3976 CrossRef CAS; (g) A. Stephenson, S. P. Argent, T. R. Johannessen, I. S. Tidmarsh and M. D. Ward, J. Am. Chem. Soc., 2011, 133, 858–870 CrossRef CAS PubMed.
- (a) A. Schaate, S. Klingelhoefer, P. Behrens and M. Wiebcke, Cryst. Growth Des., 2008, 8, 3200–3205 CrossRef CAS; (b) S. S. Kaye and J. R. Long, J. Am. Chem. Soc., 2008, 130, 806–807 CrossRef CAS PubMed; (c) K. Hirai, S. Furukawa, M. Kondo, M. Meilikhov, Y. Sakata, O. Sakata and S. Kitagawa, Chem. Commun., 2012, 48, 6472–6474 RSC.
- (a) L. Hou, J.-P. Zhang and X.-M. Chen, Cryst. Growth Des., 2009, 9, 2415–2419 CrossRef CAS; (b) S. R. Caskey, A. G. Wong-Foy and A. J. Matzger, Inorg. Chem., 2008, 47, 7751–7756 CrossRef CAS PubMed; (c) B. Chen, M. Eddaoudi, S. T. Hyde, M. O'Keeffe and O. M. Yaghi, Science, 2001, 291, 1021–1023 CrossRef CAS PubMed; (d) D. Sun, Y. Ke, T. M. Mattox, S. Parkin and H. C. Zhou, Inorg. Chem., 2006, 45, 7566–7568 CrossRef CAS PubMed.
- (a) A. Karmakar and I. Goldberg, CrystEngComm, 2011, 13, 350–366 RSC; (b) A. Karmakar and I. Goldberg, CrystEngComm, 2011, 13, 339–349 RSC; (c) A. Karmakar and I. Goldberg, CrystEngComm, 2010, 12, 4095–4100 RSC; (d) K. L. Mulfort, O. K. Farha, C. L. Stern, A. A. Sarjeant and J. T. Hupp, J. Am. Chem. Soc., 2009, 131, 3866–3868 CrossRef CAS PubMed; (e) O. K. Farha, K. L. Mulfort and J. T. Hupp, Inorg. Chem., 2008, 47, 10223–10225 CrossRef CAS PubMed; (f) X. Lin, J. Jia, X. Zhao, K. M. Thomas, A. J. Blake, G. S. Walker, N. R. Champness, P. Hubberstey and M. Schröder, Angew. Chem., Int. Ed., 2006, 45, 7358–7364 CrossRef CAS PubMed.
- (a) K. Sumida, M. L. Foo, S. Horike and J. R. Long, Eur. J. Inorg. Chem., 2010, 3739–3744 CrossRef CAS; (b) J.-P. Zhang, Y.-B. Zhang, J.-B. Lin and X.-M. Chen, Chem. Rev., 2012, 112, 1001–1033 CrossRef CAS PubMed.
- (a) S. Hasegawa, S. Horike, R. Matsuda, S. Furukawa, K. Mochizuki, Y. Kinoshita and S. Kitagawa, J. Am. Chem. Soc., 2007, 129, 2607–2614 CrossRef CAS PubMed; (b) S. Hong, Y. Zou, D. Moon and M. S. Lah, Chem. Commun., 2007, 1707–1709 RSC; (c) K. Tang, R. Yun, Z. Lu, L. Du, M. Zhang, Q. Wang and H. Liu, Cryst. Growth Des., 2013, 13, 1382–1385 CrossRef CAS; (d) B. Zheng, Z. Yang, J. Bai, Y. Lia and S. Li, Chem. Commun., 2012, 48, 7025–7027 RSC; (e) W. Zhao, J. Han, G. Tianc and X.-L. Zhao, CrystEngComm, 2013, 15, 7522–7530 RSC; (f) Y. Gong, P. G. Jiang, J. Li, T. Wu and J. H. Lin, Cryst. Growth Des., 2013, 13, 1059–1066 CrossRef CAS; (g) G.-Y. Wang, C. Song, D.-M. Kong, W.-J. Ruan, Z. Chang and Y. Li, J. Mater. Chem. A, 2014, 2, 2213–2220 RSC.
- (a) A. Karmakar, M. F. C. Guedes da Silva and A. J. L. Pombeiro, Dalton Trans., 2014, 43, 7795–7810 RSC; (b) A. Karmakar, S. Hazra, M. F. C. Guedes da Silva and A. J. L. Pombeiro, New J. Chem., 2014, 38, 4837–4846 RSC; (c) A. Karmakar, C. L. Oliver, S. Roy and L. Öhrström, Dalton Trans., 2015, 44, 10156–10165 RSC; (d) A. Karmakar, G. M. D. M. Rúbio, M. F. C. Guedes da Silva, S. Hazra and A. J. L. Pombeiro, Cryst. Growth Des., 2015, 15, 4185–4197 CrossRef CAS; (e) A. Karmakar, S. Hazra, M. F. C. Guedes da Silva, A. Paul and A. J. L. Pombeiro, CrystEngComm, 2016, 18, 1337–1349 RSC; (f) A. Karmakar, M. F. C. Guedes da Silva, S. Hazra and A. J. L. Pombeiro, New J. Chem., 2015, 39, 3004–3014 RSC; (g) A. Paul, A. Karmakar, M. F. C. Guedes da Silva and A. J. L. Pombeiro, RSC Adv., 2015, 5, 87400–87410 RSC.
- (a) A. Dhakshinamoorthy, M. Alvaro and H. Garcia, Chem. Commun., 2012, 48, 11275–11288 RSC; (b) M. Zhao, S. Ou and C.-D. Wu, Acc. Chem. Res., 2014, 47, 1199–1207 CrossRef CAS PubMed; (c) A. Corma, H. García and F. X. L. i Xamena, Chem. Rev., 2010, 110, 4606–4655 CrossRef CAS PubMed; (d) Metal–Organic Frameworks as Heterogeneous Catalysts, ed. F. L. i Xamena and J. Gascon, RSC publishing, 2013, 10.1039/9781849737586.
- (a) R. Jlassi, A. P. C. Ribeiro, M. F. C. Guedes da Silva, K. T. Mahmudov, M. N. Kopylovich, T. B. Anisimova, H. Naïli, G. A. O. Tiag and A. J. L. Pombeiro, Eur. J. Inorg. Chem., 2014, 4541–4550 CrossRef CAS; (b) R. Nasani, M. Saha, S. M. Mobin, L. M. D. R. S. Martins, A. J. L. Pombeiro, A. M. Kirillov and S. Mukhopadhyay, Dalton Trans., 2014, 43, 9944–9954 RSC; (c) A. M. Kirillov, M. V. Kirillova and A. J. L. Pombeiro, in Advances in Organometallic Chemistry and Catalysis (The Silver/Gold Jubilee ICOMC Celebratory Book), ed. A. J. L. Pombeiro, J. Wiley & Sons, 2014, 3, pp. 27–38 Search PubMed; (d) S. Hazra, A. Karmakar, M. F. C. Guedes da Silva, L'. Dlháň, R. Boča and A. J. L. Pombeiro, New J. Chem., 2015, 39, 3424–3434 RSC; (e) A. Karmakar, S. Hazra, M. F. C. Guedes da Silva and A. J. L. Pombeiro, Dalton Trans., 2015, 44, 268–280 RSC; (f) A. Karmakar, A. Paul, K. T. Mahmudov, M. F. C. Guedes da Silva and A. J. L. Pombeiro, New J. Chem., 2016, 40, 1535–1546 RSC.
- (a) J. Otera, Chem. Rev., 1993, 93, 1449–1470 CrossRef CAS; (b) J. L. Moore and T. Rovis, Carbene Catalysts, Top. Curr. Chem., 2009, 291, 118–144 CrossRef.
- (a) B. L. Salvi and N. L. Panwarb, Renewable Sustainable Energy Rev., 2012, 16, 3680–3689 CrossRef CAS; (b) F. Motasemi and F. N. Ani, Renewable Sustainable Energy Rev., 2012, 16, 4719–4733 CrossRef CAS.
- R. L. E. Furlan, E. G. Mata and O. A. Mascaretti, Tetrahedron Lett., 1998, 39, 2257–2260 CrossRef CAS.
- (a) H. Kwak, S. Hwa Lee, S. H. Kim, Y. M. Lee, E. Y. Lee, B. K. Park, E. Y. Kim, C. Kim, S.-J. Kim and Y. Kim, Eur. J. Inorg. Chem., 2008, 408–415 CrossRef CAS; (b) M. Y. Hyun, I. H. Hwang, M. M. Lee, H. Kim, K. B. Kim, C. Kim, H.-Y. Kim, Y. Kim and S.-J. Kim, Polyhedron, 2013, 53, 166–171 CrossRef CAS; (c) Y. J. Song, H. Kwak, Y. M. Lee, S. H. Kim, S. H. Lee, B. K. Park, J. Y. Jun, S. M. Yu, C. Kim, S.-J. Kim and Y. Kim, Polyhedron, 2009, 28, 1241–1252 CrossRef CAS; (d) M. Savonnet, S. Aguado, U. Ravon, D. Bazer-Bachi, V. Lecocq, N. Bats, C. Pineland and D. Farrusseng, Green Chem., 2009, 11, 1729–1732 RSC.
- (a) K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds, Wiley & Sons, New York, 5th edn, 1997 Search PubMed; (b) G. Socrates. Infrared Characteristic Group Frequencies, Wiley, New York, 1980 Search PubMed.
- (a) A. W. Addison, T. N. Rao, J. Reedijk, J. van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349–1356 RSC; (b) L. Yang, D. R. Powell and R. P. Houser, Dalton Trans., 2007, 955–964 RSC.
- (a) M. O'Keeffe and O. M. Yaghi, Reticular Chemistry Structure Resource, Arizona State University, Tempe, AZ, 2005, http://www.rcsr.anu.edu.au/ Search PubMed; (b) V. A. Blatov, IUCr, Compcomm Newsletter, 2006, 7, 4 Search PubMed; (c) V. A. Blatov, Struct. Chem., 2012, 23, 955–963 CrossRef CAS.
- R. A. Sheldon, M. Wallau, I. W. C. E. Arends and U. Schuchardt, Acc. Chem. Res., 1998, 31, 485–493 CrossRef CAS.
- (a) Bruker, APEX2 & SAINT, AXS Inc., Madison, WI, 2004 Search PubMed; (b) G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed; (c) L. J. Farrugia, J. Appl. Crystallogr., 1999, 32, 837–838 CrossRef CAS; (d) A. L. Spek, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, C34 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1490728–1490732. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra17518j |
| This journal is © The Royal Society of Chemistry 2016 |