
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConsiderable Fenton and photo-Fenton reactivity of passivated zero-valent iron†
Marco Minellaa,
Elisabetta Sappaa,
Khalil Hanna*b,
Francesco Barsottia,
Valter Maurinoa,
Claudio Mineroa and
Davide Vione*a
aDipartimento di Chimica, Università di Torino, Via Pietro Giuria 5, 10125 Torino, Italy. E-mail: davide.vione@unito.it; Fax: +39-011-6705242; Tel: +39-011-6705296
bEcole Nationale Supérieure de Chimie de Rennes, CNRS, UMR 6226, 11 Allée de Beaulieu, CS 50837, 35708 Rennes Cedex 7, France. E-mail: khalil.hanna@ensc-rennes.fr; Fax: +33-02-23238120; Tel: +33-02-23238027
First published on 31st August 2016
Abstract
The vulnerability of Zero-Valent Iron (ZVI) to passivation, which significantly decreases its surface reactivity, limits its use as a reducing agent in environmental remediation applications (e.g., permeable reactive barriers). Passivation of ZVI occurs rapidly in ambient environments due to the presence of oxygen and water, as well as during the reduction of contaminants (e.g. chlorinated and nitrated organic compounds) even in anoxic conditions. Passivated zero-valent iron (pZVI) particles are typically covered by an iron (hydr)oxide layer that entails a loss of the typical properties of this material, prevents the reductive activity to take place, and could made pZVI a waste to be disposed of. This work shows that pZVI has considerable Fenton (oxidizing) reactivity in the presence of H2O2 at acidic pH. The pZVI-Fenton process likely involves reaction between H2O2 and the pZVI surface or near-surface species, because the detected levels of leached Fe in the solution bulk were insufficient to account for the observed degradation processes. Still, leached Fe in the solution at pH 2 and 3 would be too concentrated for some possible applications (e.g. wastewater treatment, which would require an additional precipitation step to remove excess Fe). In such a case, operation at pH 4 would be preferred to maintain reasonable reactivity without measurable Fe leaching. The pZVI-Fenton process was inactive above pH 4, but reactivity could be extended up to pH 5 by UVA irradiation. Surprisingly, a pZVI sample that underwent an additional 78 day aging in aqueous solution exhibited a considerable ability to photochemically remove phenol despite a slower initial stage. These properties account for the possible use of a potential waste in decontamination reactions.
1. Introduction
Zero-valent iron (ZVI) is a strong reducing agent widely used for the reductive removal of different organic (chlorinated and nitroaromatic compounds), inorganic (chromate, arsenite and nitrate), and radioactive (uranium(VI)) pollutants.1–16 ZVI is used for groundwater remediation in the form of permeable reaction barriers,17,18 or as a slurry to be injected into the contaminated aquifer.19 In the latter case, the use of a suitable dispersing agent is very important to increase the mobility of ZVI particles in the porous medium.20,21 Recently, ZVI has also been proposed as a reactant for the decontamination of wastewater, where it finds application towards the same contaminants that are targeted in groundwater remediation.18,22 There is growing use of ZVI technology in environmental remediation processes, due to its high reductive capacity, low cost when compared with other zero-valent metallic particles (e.g. bimetallic Ni–, Pd–, Cu–, Co– and Au–Fe0), the environmental friendliness of iron, and the production of non-toxic by-products.5,18Although ZVI technologies have been extensively studied for the reduction of environmental contaminants, the rapid passivation of the ZVI surface considerably limits possible applications. In fact, ZVI particles due to their reducing power undergo a rapid surface oxidation by oxygen, as well as in O2-free aqueous solutions. Indeed, the anaerobic corrosion of ZVI in water produces ferrous ions (eqn (1)), whereas the aerobic corrosion generates ferrous ions and OH− (eqn (2)), or ferrous ions and H2O2 at acidic pH (eqn (3)).23,24
| Fe0 + 2H2O → FeII + 2OH− + H2 | (1) |
| Fe0 + ½O2 + H2O → FeII + 2OH− | (2) |
| Fe0 + O2 + 2H+ → FeII + H2O2 | (3) |
A further formation of H2O2 occurs via the oxidation of FeII by O2 (eqn (4) and (5)), mainly at pH values above 6.25
| FeII + O2 → FeIII + O2−˙ | (4) |
| FeII + O2−˙ + 2H+ → FeIII + H2O2 | (5) |
The generated H2O2 can oxidize ZVI to give ferrous ions (eqn (6)) or react directly with FeII species to promote the production of OH˙ at acidic pH according to the classic thermal Fenton reaction (eqn (7)), whereas an important formation of superoxidized iron species, such as the ferryl ion, is expected in neutral to basic conditions (eqn (8)).26 These reactions are often utilized to explain the oxidative transformation of contaminants observed in the presence of oxygen and nano-scale ZVI.27,28
| Fe0 + H2O2 + 2H+ → FeII + 2H2O | (6) |
| FeII + H2O2 → FeIIIOH2+ + OH˙ | (7) |
| FeII + H2O2 → FeIVO2+ + H2O | (8) |
The surface oxidation of Fe0 is also observed during the reduction of contaminants even in anoxic environments,29,30 as for instance for nitro-compounds (eqn (9)) or halogenated compounds (eqn (10), where X is a halogen atom):
| 3Fe0 + R–NO2 + 6H+ → 3FeII + R–NH2 + 2H2O | (9) |
| Fe0 + RX + H+ → FeII + RH + X− | (10) |
According to reactions (1–7), precipitates composed of mixed FeII–FeIII and/or FeIII species may be formed as a coating layer of ZVI leading to the passivation of the surface. These species cover the surface of ZVI particles and prevent Fe0 from carrying out its typical reactions.31,32 For this reason, surface passivation becomes one of the most important issues to be overcome in order to increase the longevity or reactivity of ZVI in environmental contaminated systems. A possible approach is the depassivation/dissolution of the oxide layer coating the aged Fe0, which could for instance take place in the presence of some salts (Cu2+, Co2+, Mn2+, Mg2+, Pb2+, Ni2+) and could have implications for the treatment of contaminated water in marine or otherwise highly saline environments.33–35 If, on the contrary, it is not possible to achieve depassivation, ZVI becomes unusable and a potential waste to be disposed of. This problem is relevant both to groundwater remediation, where a fast ZVI recycling is difficult and gradual passivation is almost unavoidable in normal operations,36 and to wastewater remediation, where passivation places a limit to the use of ZVI in repeated cycles.22,37 In this paper we show that passivated ZVI (hereafter, pZVI) shows important Fenton and photo-Fenton reactivity in the presence of H2O2. The added oxidant (H2O2) could react with FeII of the oxide coating layer of ZVI to generate reactive oxygen species, or with FeIII via the Haber–Weiss cycle. This finding opens up a new strategy to employ ZVI in remediation processes, in alternative to depassivation: fresh ZVI could be used in reduction reactions while passivated ZVI, activated upon addition of H2O2, could find application in oxidative steps.
In this work, the ability of pZVI to promote Fenton and photo-Fenton reactions was assessed by using phenol as model compound because: (i) phenol is a highly studied substrate with well-known behavior in advanced oxidation processes, which makes it a first choice in the study of the oxidative reactivity of new or poorly known materials;38–40 (ii) phenol is an important component of oil refinery wastes and a by-product of the conversion of coal into gaseous or liquid fuels and of the manufacturing of metallurgical coke from coal. Therefore, phenol can occur in the environment and particularly in environmental waters because of discharges of oil refineries, coal conversion plants, municipal waste treatment plants, and spills. Phenol is highly toxic to both humans and animals, and its removal by conventional physico-chemical and biological technologies is often incomplete.41,42 This issue accounts for the need to investigate on effective methods for phenol degradation.
2. Experimental section
2.1. Reagents and materials
Phenol (purity grade 99%), 2-chlorophenol (99%), 4-chlorophenol (99%), sodium citrate (98%), 4-aminoantipyrine (reagent grade), methanol (gradient grade), H3PO4 (85%), HClO4 (70%), NaCl (99.5%) and NaH2PO4·H2O (98%) were purchased from Aldrich, horseradish peroxidase from Sigma, sodium oxalate (99.5%), FeSO4·7H2O (99.5%), Fe(NO3)3·9H2O (98%) and H2SO4 (96%) from Carlo Erba, H2O2 (35%), Na2S2O8 (98%), KSCN (99%) and Na2HPO4·2H2O (99.5%) from VWR Int. These reagents were used as received, without further purification. Water used was of Milli-Q quality.Passivated zero-valent iron (pZVI) was obtained by exposing a commercial ZVI sample (Fe0 powder, 99%, ∼70 mesh, <212 μm, Acros Organics) to air over around three months, following the procedure reported in Liu et al.34 This ageing procedure passivates ZVI with a layer of mixed oxides and thus quenches its reactivity.34 In addition, some pZVI samples were further aged in aerated solutions for a variable time, which changed the visual appearance of the aqueous slurries (see below). Before and after the ageing in aerated aqueous suspension, pZVI was firstly analyzed by X-ray diffraction (XRD) with a D8 BRUKER diffractometer, and then characterized by transmission electron microscopy with selected area diffraction (TEM-SAD) (JEM-2100, JEOL).
X-ray diffraction (XRD) analysis confirmed that the pZVI sample was essentially Fe0, with a minor presence of Fe3O4 (magnetite) and/or γ-Fe2O3 (maghemite). Indeed, XRD data of pZVI particles (Fig. 1) revealed the presence of main peaks of α-Fe (44.6°, 65° and 82° 2θ), and magnetite or maghemite (35.3°, 42° and 57° 2θ). The XRD technique is unfortunately not very suitable for quantification purposes, in particular as far as the composition of the surface layer is concerned. As an alternative technique the X-ray photoelectron spectroscopy (XPS), although able to analyze the surfaces of solids, is not really suitable for a quantification (even a semi-quantitative assessment) of FeII and FeIII species. Therefore, only a rough assessment of the pZVI surface species could be obtained here.
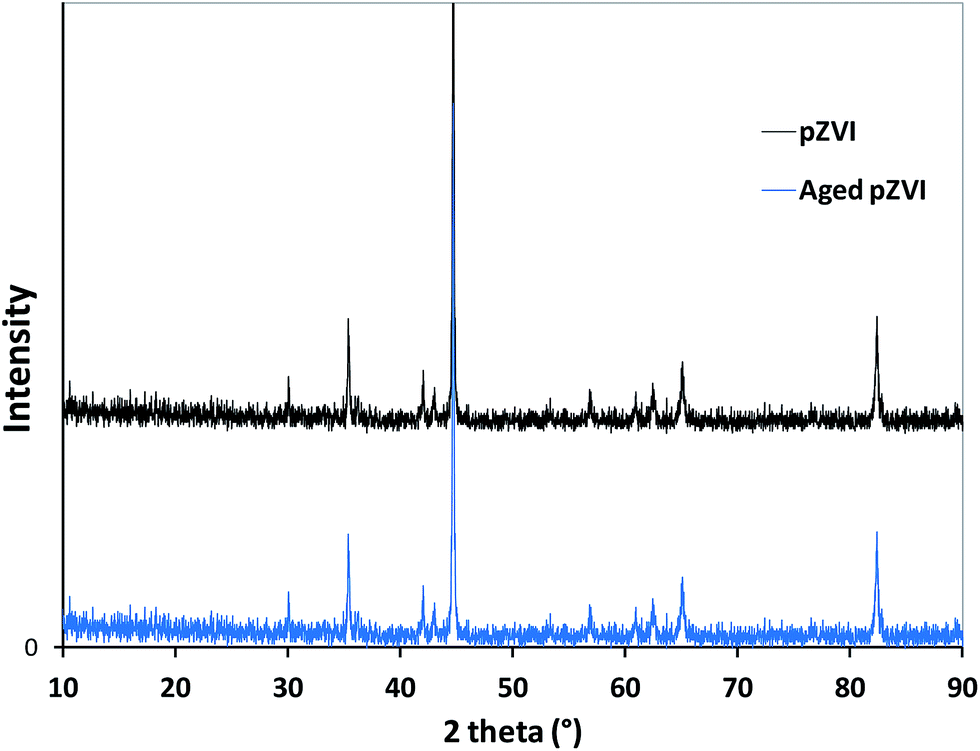 | ||
| Fig. 1 XRD diffractograms showing the pZVI before and after ageing in O2-containing aqueous suspension for one month. | ||
Transmission electron micrographs (TEM) images of the pZVI particles (Fig. 2) suggested a wide particle size distribution, i.e. very small spherical particles (∼20 nm) next to bigger and shapeless particles (∼2 μm). This finding agrees with the results of particle size distribution analysis by laser diffraction performed on the pristine commercial ZVI sample, which has suggested a very broad particle heterogeneity with an average size of around 167 μm.34
 | ||
| Fig. 2 TEM images showing the initial pZVI (a–c) that underwent passivation in air and aged pZVI (d–f) that underwent additional ageing for one month in aerated suspension. | ||
2.2. Phenol degradation experiments
The solutions containing phenol (100 mL total volume) were placed in 200 mL beakers and were mechanically stirred. Depending on the goal of each experiment, the system could also contain pZVI and H2O2. The pH values were adjusted with HClO4. The choice of this acid is motivated by the fact that the perchlorate anion does not react significantly with ˙OH, thus ensuring the absence of biases in Fenton experiments. In contrast, the anions of other mineral acids such as chloride and sulfate react with ˙OH in acidic solution (sulfate does so below pH 2 because of the occurrence of HSO4−), while nitrate is a ˙OH source under irradiation.43 In some runs in the dark the Fe source was dissolved FeSO4 or Fe(NO3)3 instead of pZVI. In some experiments the beaker was irradiated with a UVA black lamp (Philips TL-D 18 W, emission maximum at 368 nm), producing an irradiance of 20.5 W m−2 on top of the irradiated systems. In all the cases, sample aliquots were taken at scheduled times from the reaction mixture and pZVI was removed by syringe filtration, using Millipore Millex HV filters (0.45 μm pore diameter). For the monitoring of phenol, 0.5 mL of the clear (filtered) solution were added to an HPLC vial that contained 0.5 mL methanol, to quench possible residual Fenton processes that might still be operational in the homogeneous phase. Analysis was carried out by liquid chromatography with diode array detection (HPLC-DAD), using a VWR-Hitachi Elite LaChrom instrument equipped with L2200 autosampler (sample volume 60 μL), L2130 quaternary pump module for low-pressure gradients, Duratec vacuum degasser, L2300 column oven (set at 40 °C), and L2455 diode-array detector. The samples were eluted with a 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :70 mixture of methanol and aqueous H3PO4 (pH 2.8) at 1.0 mL min−1 flow rate, using a column Merck LiChroCART packed with LiChrospher 100 RP18 (125 mm × 4 mm × 5 μm). Phenol (retention time 5.7 min, column dead time 1.2 min) was monitored at 210 nm. The repeatability of replicate experiments was about 10–20%. Error bars are not shown on the plots for readability issues.
:70 mixture of methanol and aqueous H3PO4 (pH 2.8) at 1.0 mL min−1 flow rate, using a column Merck LiChroCART packed with LiChrospher 100 RP18 (125 mm × 4 mm × 5 μm). Phenol (retention time 5.7 min, column dead time 1.2 min) was monitored at 210 nm. The repeatability of replicate experiments was about 10–20%. Error bars are not shown on the plots for readability issues.
2.3. Monitoring of dissolved Fe, H2O2 and total organic carbon (TOC)
In these determinations, methanol was not used as reaction quencher to avoid analytical biases. Therefore, care was taken to carry out the analyses soon after sampling. Dissolved Fe and its speciation were determined spectrophotometrically,44 using a Varian Cary 100 Scan double-beam, UV-Vis spectrophotometer and Hellma quartz cuvettes (1 cm optical path length). At selected reaction times, 15 mL sample aliquots were withdrawn from the reaction mixture and syringe filtered as mentioned above. For the determination of total Fe, a 5 mL sub-aliquot was placed in a 10 mL flask and added with aqueous solutions of K2S2O8 (0.5 g L−1, 1 mL), H2SO4 (0.1 M, 1 mL), KSCN (0.5 g L−1, 1 mL), and with water to volume. In this way, dissolved FeII was oxidized to FeIII by S2O82−, and total Fe could be quantified as FeSCN2+ at 457 nm. Fe(III) was determined on a further 5 mL sub-aliquot in a similar way, but without addition of K2S2O8. The concentration of FeII was obtained as the difference between total Fe and FeIII. Under these conditions, the Fe detection limit was 0.05 mg Fe L−1.Hydrogen peroxide was also determined spectrophotometrically, using the peroxidase–4-aminoantipyrine method.45 The color-forming reagent was prepared by dissolving 0.10 g 4-aminoantipyrine, 0.23 g phenol, 1 mg horseradish peroxidase, 0.71 g NaH2PO4 and 0.28 g Na2HPO4 in 100 mL water. Sample aliquots for H2O2 monitoring (10 mL) were withdrawn from the reaction mixture and filtered. In each 10 mL flask it was placed a filtered sample sub-aliquot of 0.5 to 5 mL, 4 mL reagent and water to volume. The volume of filtered sample added in each case was determined with the aims of carrying out the measure within the linearity range of the method (0.05–0.25 mM H2O2) and of maximizing the sensitivity of the whole analytical procedure, avoiding unnecessary dilution of the sample. The colored adduct between oxidized phenol and 4-aminoantipyrine was detected at 505 nm.
To monitor the TOC, 15 mL sample aliquots were withdrawn at scheduled times, filtered and injected into a Shimadzu TOC-V-CSH total organic carbon analyzer. The TOC was determined as the difference between total carbon and inorganic carbon.
3. Results and discussion
The inability of the pZVI material to act as an effective reducing agent was tested towards the dechlorination of 2- and 4-chlorophenol, which yield phenol and chloride ions upon reduction in the presence of ZVI.46–49 Attempts aimed at achieving surface depassivation, by addition of ligands for Fe ions such as oxalate and citrate, or with a combination of ligands and irradiation (with the purpose of dissolving the iron oxide layer50 and allowing the underlying Fe0 to get in contact with the solution) were not successful and no dechlorination of either 2- or 4-chlorophenol was observed. However, pZVI proved to be effective in inducing the Fenton reaction in the presence of H2O2. The rest of this work is devoted to the study of the Fenton and photo-Fenton reactivity of pZVI under a variety of conditions.3.1. Fenton reactivity of pZVI
The effect of pH on the Fenton degradation of phenol induced by pZVI is shown in Fig. 3. The process was fastest at pH 3 and became ineffective at pH ≥ 5. Interestingly, in the optimal pH conditions a complete disappearance of phenol was observed in less than 10 min reaction time. The reported pH trend is not much different from that observed in the homogeneous phase,57,58 but it does not make conclusive evidence in favor of the site(s) where the Fenton reaction takes place (involving either dissolved Fe or Fe-sites on or near the pZVI surface, vide infra).
 | ||
| Fig. 3 Time trend of phenol (initial concentration 0.1 mM) in the presence of 1 mM H2O2 and 0.1 g L−1 pZVI at different pH values, adjusted by addition of HClO4. The error bars are not reported on this or other plots for a sake of readability. The reproducibility among repeated experiments was in the range of 10–20%. | ||
 | ||
| Fig. 4 Time trend of phenol (initial concentration 0.1 mM) in the presence of 0.1 g L−1 pZVI at pH 3, adjusted by addition of HClO4, with different concentration values of H2O2. | ||
In contrast, phenol was degraded in the presence of pZVI + H2O2 (Fig. 4) and the degradation was most efficient for a H2O2 concentration level around 1 mM. Lower H2O2 values would reduce the rate by which reactive oxidizing species are generated, while higher concentrations would entail a significant scavenging of reactive species by H2O2 itself (hydrogen peroxide is able to scavenge ˙OH with a second-order reaction rate constant of 2.7 × 107 M−1 s−1).61
The 1 mM H2O2 concentration that induces very fast and complete removal of 0.1 mM phenol is stoichiometrically insufficient to achieve total mineralization, because the transformation of a phenol molecule into six CO2 molecules requires the abstraction of 28 electrons. Since H2O2 is a 2-electron oxidant, the mineralization of a phenol molecule would require at least 14H2O2. Therefore, the 10:1 H2O2:phenol ratio used here is not enough to mineralize phenol if H2O2 is the only oxidant species. Such a scenario is modified by the fact that H2O2 is activated to produce ˙OH and that additional redox reactions between radical species (formed from phenol + ˙OH) and dissolved O2 would also contribute to the oxidation process.62 The time trend of the TOC was monitored at pH 3 in the presence of 0.1 mM phenol (corresponding to an initial TOC of 7.2 mg C L−1), of 0.1 g L−1 pZVI and, in separate experiments, of 1, 3.5 and 10 mM H2O2. In the case of 1 mM H2O2, the TOC decreased by around 2 mg C L−1 (corresponding to 25–30% of mineralization) in 1 h reaction time and then remained approximately constant. The observed degree of mineralization was considerably lower than the theoretical value of ∼70% that would be obtained by purely stoichiometric considerations. Insignificant mineralization was then observed with 3.5 and 10 mM H2O2, thereby suggesting that the improvement of the oxidation stoichiometry (due to a higher H2O2:phenol ratio) was largely offset by the scavenging of reactive species carried out by H2O2.
Overall, the Fenton reaction catalyzed by pZVI appears to be a potentially effective way to induce the degradation of the primary substrate, but not to achieve total mineralization. However, in several practical applications the pollutants occur in the aqueous phase together with dissolved organic matter (DOM), and the target of total mineralization would also entail the unnecessary transformation of DOM into CO2. In such a case, the selective mineralization of the pollutant(s) could be non attainable or, in any case, not experimentally verifiable. Still, in the absence of mineralization one should take into account the potential health effects and environmental fate of the reaction mixture.63 The case of phenol is not problematic because it is not difficult to avoid the formation of possibly toxic intermediates by controlling the Fenton operational conditions.64 Moreover, the biodegradability of xenobiotics often increases significantly during the first oxidative (Fenton) steps, although mineralization may not be achieved, and a combined process with a biological post-treatment is an interesting option.65 However, the harmful effects of the transformation intermediates may be very important for some pollutants, and the suitability of pZVI-Fenton as treatment system should be checked case by case, depending on the substrate to be degraded.
The experiments were carried out at several values of pH and H2O2 concentration, in the presence of 0.1 mM phenol and 0.1 g L−1 pZVI. Fig. 5A and B reports the results obtained at pH 2, with H2O2 1 mM (Fig. 5A) and 3.5 mM (Fig. 5B). In the former case, H2O2 concentration halved very quickly and hydrogen peroxide became undetectable after 20 min. The FeIII concentration peaked at around 10 min reaction time, while FeII increased up to 45 min. The initial strong prevalence of FeIII over FeII is accounted for by the occurrence of residual H2O2, which oxidizes FeII to FeIII (reaction (7)). The FeII oxidation process is expected to stop after the disappearance of H2O2, coherently with the time trend of the Fe species. Fig. 5B shows that 3.5 mM H2O2 took longer to disappear from the system (∼90 min) than 1 mM H2O2 (∼20 min). Coherently, with 3.5 mM H2O2 the FeIII levels were initially much higher than those of FeII and started decreasing only after ∼90 min reaction time.
 | ||
| Fig. 5 Time trends of FeIII, FeII (left Y-axis) and H2O2 (right Y-axis), in the presence of 0.1 mM phenol, 0.1 g L−1 pZVI, and: (A) 1 mM H2O2 at pH 2; (B) 3.5 mM H2O2 at pH 2; (C) 1 mM H2O2 at pH 3; (D) 3.5 mM H2O2 at pH 3. The pH values were adjusted by addition of HClO4. | ||
At pH 3 the concentration of dissolved FeIII was lower than at pH 2 (see Fig. 5C and D), which is consistent with a decrease of the FeIII solubility with increasing pH.55,56 In the presence of 1 mM H2O2 at pH 3 (Fig. 5C), FeIII was the main dissolved Fe species for reaction times <20 min, when H2O2 was still detectable. Moreover, the build-up of FeII with time is consistent with the disappearance of H2O2. The decrease of dissolved FeIII after 10 min reaction time could be due to precipitation on the solid, reduction to FeII, or both. In the case of 3.5 mM H2O2, which became undetectable after 300 min, the level of dissolved FeIII was higher than that of FeII until H2O2 disappearance (Fig. 5D).
At pH 4 or higher, dissolved Fe could not be detected in either oxidation state, which is again consistent with a decreasing solubility of the Fe species with increasing pH. At pH 4, phenol disappeared within 30 min reaction time in the presence of pZVI + 1 mM H2O2 (see Fig. 3), thereby suggesting that the reaction took place independently of the occurrence of dissolved Fe. The latter inference is further supported by the fact that phenol disappeared in about 40 min in the presence of 3.5 mM H2O2 at pH 3 (see Fig. 4), at which reaction time the dissolved Fe was still below detection limit (see Fig. 5D). These findings suggest that a homogeneous-phase reaction between H2O2 and dissolved Fe may not be necessary to trigger the degradation of phenol. In contrast, a reaction of H2O2 with the pZVI surface should be invoked to account for the observed results.
To further stress the above point, phenol degradation was studied in the presence of 1 mM H2O2 at pH 2 and 3 in the homogeneous phase, without pZVI but with comparable levels of dissolved Fe as those detected in the pZVI runs (see Fig. 1 in the ESI;† homogeneous Fe was obtained by dissolution of FeSO4 or Fe(NO3)3). The homogeneous-phase experiment at pH 3 was carried out in the presence of 6 mg L−1 dissolved FeIII, because in the corresponding pZVI run the dissolved FeII was still undetectable by the time phenol underwent complete degradation. The results of the homogeneous-phase experiments showed that phenol degradation with dissolved Fe + H2O2 was always considerably slower compared to pZVI + H2O2. Therefore, it is suggested that the pZVI-Fenton process involves reaction between H2O2 and either the pZVI surface or iron species located in the interfacial double layer of the colloidal particles, to a higher extent than reaction of H2O2 with dissolved Fe species in the solution bulk.
Coming back to the results shown in Fig. 5, it appears that dissolved Fe in the pZVI runs at pH 2 and 3 easily reached levels higher than 1 mg Fe L−1. Therefore, in contexts where dissolved Fe levels above 1 mg Fe L−1 may be a problem (e.g. for discharge of wastewater into receiving water bodies), operation at pH 4 that gave leached Fe <0.05 mg L−1 could be a good compromise to achieve pollutant degradation with limited Fe release.
Given the higher reactivity of nanosized minerals,73 we can suppose that a rapid oxidation of very small particles may first occur in our pZVI suspensions having a broad particle size distribution. This is confirmed by TEM analysis of the water-aged sample, where we can observe some needles (acicular-like particles) (Fig. 2) which may correspond to lepidocrocite or goethite that generally have this form in aqueous suspension.74 However, formation of FeIIIOOH could not be identified by XRD (Fig. 1), probably because of its very low amount with respect to the bulk metallic phase (α-Fe). Indeed, the proportion of very small particles is supposed to be relatively low according to the particle size analysis.34
These observations suggest that particles in pZVI suspensions may have different behaviors towards oxidation, and that ageing in aqueous suspension (under open air atmosphere) may essentially oxidize the smallest particles (the most reactive ones) to FeIIIOOH, as previously observed for nano-sized ZVI particles.67 This may modify the surface charge of particles in pZVI suspensions, thereby altering the electrostatic interactions between the different kinds of particles, as well as the aggregation state in the suspensions (as previously mentioned, see Fig. 2 ESI†).
The influence of the storage time on the Fenton reactivity of pZVI was tested at different pH values, using phenol as model molecule. Fig. 6 shows the time trend of 0.1 mM phenol in the presence of 0.1 g L−1 pZVI and 1 mM H2O2, at pH 2 (Fig. 6A), 4 (Fig. 6B) and 6 (Fig. 6C). A test at pH 3 is not reported, because the elevated pZVI reactivity caused very fast degradation of phenol and made it very difficult to highlight small differences between degradation experiments.
 | ||
| Fig. 6 Time trend of phenol (initial concentration 0.1 mM) in the presence of 1 mM H2O2 and 0.1 g L−1 pZVI. The pZVI stock aqueous suspension (1 g L−1 loading) was stored in the presence of air for the reported amount of time prior to use. (A) pH 2; (B) pH 4; (C) pH 6. The pH values were adjusted by addition of HClO4. | ||
The runs at pH 2 did not show a trend as a function of the ageing time. Small differences between experiments are likely accounted for by random variability, independently of ageing. A similar issue was observed at pH 4; finally, insignificant phenol degradation took place at pH 6 in the presence of pZVI and H2O2. The runs at pH 6, where pZVI was practically non-reactive, give insight into the overall variability concerning the monitoring of phenol concentration. These results strengthen the interpretation given to the data obtained at pH 2 and 4, namely that the limited variations observed in different experiments were not due to ageing but to experimental repeatability issues.
The Fenton reactivity of pZVI was thus not affected by the ageing of particles in O2-containing water. While the ageing process may entirely transform nanosized particles into FeIII-oxides in aerated water,67 only an outer coating layer may be formed on microsized ZVI surfaces upon oxidation. Experimental evidences of these mineralogical transformations are hard to obtain, particularly in pZVI suspension with broad particle size heterogeneity. The oxidation by-products of ZVI or the passivated layer formed on ZVI surfaces are generally composed of magnetite, maghemite, goethite, lepidocrocite and/or hematite.67–72 The latter may have different reactivities for the Fenton reaction, as the FeII–FeIII mixed valence oxides are known to be more effective than only FeIII oxides,75 which are expected to be formed preferentially from the smaller particles.67 In addition, the FeII/FeIII ratio in mixed oxides considerably affects the intrinsic reactivity for the Fenton or photo-Fenton oxidation.39,76 Therefore, it is possible that different processes taking place during ageing compensate for each other as far as the Fenton reactivity of pZVI is concerned.
3.2. Photo-Fenton reactivity of pZVI at higher pH values under UVA irradiation
Phenol degradation by pZVI + H2O2 was also studied under irradiation. The dark Fenton reactivity of pZVI at pH 4 or lower is obviously a confounding factor for the irradiation experiments, which were thus carried out at pH 5 or higher where the dark reactions are not operational. Fig. 7 shows the time trend of phenol at pH 5 (by HClO4) in the presence of pZVI + H2O2 in the dark, of H2O2 under irradiation, and of pZVI + H2O2 under irradiation. In the latter case, two pZVI suspensions were tested: one was freshly prepared, and the other was aged for 78 days before use. As already explained, ageing of pZVI took place in an air-equilibrated suspension. No dark reactivity could be highlighted at pH 5, as expected, while the limited phenol degradation with irradiated H2O2 (without pZVI) would be accounted for by the direct photolysis of hydrogen peroxide to give ˙OH.77,78 Significant phenol degradation was achieved with pZVI + H2O2 under irradiation, with an interesting difference between the two pZVI suspensions. Degradation was initially faster with the freshly prepared pZVI, but complete phenol disappearance was not achieved. In contrast, total phenol removal was obtained with the 78 d-aged pZVI suspension, despite the slower initial degradation stage. | ||
| Fig. 7 Time trend of phenol (initial concentration 0.1 mM) in the presence of 1 mM H2O2 and, according to the different experiments (see figure legend): (■) fresh pZVI (0.1 g L−1) in the dark (“fresh pZVI” denotes the material that underwent initial ageing in air but did not undergo additional ageing in aqueous suspension); (●) fresh pZVI (0.1 g L−1) under UVA irradiation; (▲) aged pZVI (0.1 g L−1) under UVA irradiation (this material was aged in aqueous suspension for 78 days), and (▼) no pZVI under UVA irradiation. In all the experiments the pH value was 5, adjusted with HClO4. | ||
The reactivity of pZVI under irradiation in the presence of H2O2 could be due to three main factors: (i) the photolysis of ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) FeIII–OH surface groups to produce ˙OH and FeII; (ii) the photolysis of H2O2 and, last but not least, (iii) the Fenton reaction involving either surface-bound or dissolved FeII. Due to the low solubility of Fe-oxides and low release of Fe ions at high pH values, no dissolved Fe (either ferrous or ferric ion) was detected in pZVI suspensions at pH 5.
FeIII–OH surface groups to produce ˙OH and FeII; (ii) the photolysis of H2O2 and, last but not least, (iii) the Fenton reaction involving either surface-bound or dissolved FeII. Due to the low solubility of Fe-oxides and low release of Fe ions at high pH values, no dissolved Fe (either ferrous or ferric ion) was detected in pZVI suspensions at pH 5.
When considering the different behavior of the two pZVI samples, we can suppose that the availability and reactivity of Fe-surface groups can vary with the ageing time in aqueous suspension.79 Indeed, the reaction shows a short initial lag-phase in the 78 d-aged pZVI suspension, but with higher eventual removal compared to the fresh pZVI suspension. As explained above, ageing in aerated water may mainly oxidize the smallest particles (the most reactive) to FeIII–oxihydroxides, and then modify the aggregation state (i.e. exposed surface area of particles) in pZVI suspensions. This modification might induce changes in the availability of FeIII–OH surface sites in suspension, which are potential photoactive groups under UVA irradiation.43 If we make the reasonable hypothesis that the fresh pZVI surface contains a mixture of FeII and FeIII, while aged pZVI is more extensively oxidized with a prevalence of FeIII groups, the fact that aged pZVI induced complete photooxidation of phenol could be accounted for by the occurrence of a higher amount of photoactive FeIII compared to fresh pZVI, while FeII (higher in fresh pZVI) is ineffective in the presence of H2O2 at pH 5. The initially higher photoreactivity of fresh pZVI could be linked to the fact that the occurrence of FeII produces a magnetite-like surface, and magnetite shows a higher activity under photo-Fenton conditions at ∼neutral pH compared to a pure FeIII phase (e.g. hematite).39,80
Irradiation of phenol with pZVI + H2O2 was also carried out at pH 6, and negligible degradation could be observed under such conditions. Compared to the absence of degradation with irradiated pZVI + H2O2 at pH 6, some phenol degradation at the same pH was observed with irradiated H2O2 alone, without pZVI (∼20% of the initial phenol was degraded in 24 h). The most likely explanation is that, at pH 6, inactive pZVI just shields radiation that is no longer available for H2O2 photolysis. It can thus be concluded that pZVI is inactive in the dark Fenton process above pH 4, and photochemically inactive above pH 5. The extension towards higher pH values of the applicability of the photo-Fenton reaction, compared to the dark Fenton process, is consistent with previous reports.81
4. Conclusions
The pZVI sample studied in this work had lost, due to passivation, its reduction capability as tested towards the dechlorination of chlorophenols. In contrast, it had significant Fenton and photo-Fenton (oxidizing) reactivity in the presence of H2O2. The dark pZVI-Fenton process, tested against the degradation of phenol, was most effective at pH 3 but it was no longer operational at pH 5 or higher. The presence of H2O2 was essential to trigger reactivity up to an optimum concentration (1 mM H2O2 in the presence of 0.1 mM phenol and 0.1 g L−1 pZVI at pH 3). Above the optimum H2O2 concentration the degradation of phenol became gradually less effective, presumably because of the scavenging of reactive species by H2O2 itself.The pZVI-Fenton reaction occurred along with the dissolution of Fe species at pH < 4. In particular, FeIII prevailed in the dissolved phase until H2O2 was totally consumed, while the H2O2 disappearance allowed the build-up of FeII. Despite phenol degradation, no dissolved Fe was detected at pH 4. The levels of dissolved Fe were never high enough to account for an important homogeneous Fenton process between the aqueous-phase Fe species and H2O2. Therefore, it is hypothesized that most of phenol would be degraded either by an heterogeneous reaction between H2O2 and pZVI particles or immediately in the proximity of the particle surface, by the reaction of H2O2 with Fe2+ ions located in the interfacial double layer of the colloidal particles. The detected levels of dissolved Fe at pH 2 and 3 were higher than 1 mg L−1, which would be a problem in some possible applications such as wastewater treatment. In these cases, the operation at pH 4 would be the best compromise between the possibility to degrade phenol and the minimization of Fe leaching. Alternatively, the use of pZVI-Fenton at pH < 4 would require additional steps to remove the excess of iron (e.g. pH adjustment and addition of flocculation agents).
Although an effective degradation of phenol could be achieved under several different conditions, the pZVI-Fenton system was not able to attain total mineralization. The best results (25–30% mineralization of 0.1 mM phenol) were obtained with 1 mM H2O2 at pH 3. An increase of H2O2 concentration, which in theory could induce a more favorable stoichiometric ratio between the oxidant and phenol, was actually detrimental. The most likely reason is the scavenging of reactive species by H2O2, which decreases the reactivity of the system.
The ageing time in aerated suspension, i.e. an additional forced passivation of pZVI particles in O2-containing water, did not affect the Fenton reaction performance. This remarkable catalytic stability of pZVI for a wide range of ageing times has strong implications for field applications, particularly where the iron slurry is injected in a groundwater portion (or engineering reactor) for a certain time.
The dark pZVI-Fenton process was inactive above pH 4. Reactivity could be extended to pH 5 upon UVA irradiation of the system, probably because of additional phenomena such as photolysis of Fe(III) (hydr)oxides and of H2O2. Finally, no Fenton or photo-Fenton reactivity was observed at pH 6 or higher. Surprisingly, long term-aged ZVI exhibited a high photo-Fenton reactivity and total removal of phenol in aqueous suspension at pH 5.
The data presented here show that pZVI, which due to inactivity in reductive processes is an inert material and possibly a waste to be disposed of, can be re-used in Fenton oxidation reactions upon addition of H2O2. Therefore, ZVI can be employed as a reductant until it retains sufficient reactivity, and then be converted into a catalyst for oxidative processes in the presence of H2O2. The possibility to reuse a waste makes pZVI a potentially very cheap Fenton reactant.
References
- F. He, D. Y. Zhao and C. Paul, Water Res., 2010, 44, 2360–2370 CrossRef CAS PubMed.
- B. Lai, Y. H. Zhang, R. Li, Y. X. Zhou and J. L. Wang, Chem. Eng. J., 2014, 249, 143–152 CrossRef CAS.
- P. Mueller, K. E. Lorber, R. Mischitz and C. Weiss, Sci. Total Environ., 2014, 485, 748–754 CrossRef PubMed.
- S. Klas and D. W. Kirk, J. Hazard. Mater., 2013, 252, 77–82 CrossRef PubMed.
- F. L. Fu, D. D. Dionysiou and H. Liu, J. Hazard. Mater., 2014, 267, 194–205 CrossRef CAS PubMed.
- C. B. Wenk, R. Kaegi and S. J. Hug, Environ. Chem., 2014, 11, 547–557 CrossRef CAS.
- M. J. Alowitz and M. M. Scherer, Environ. Sci. Technol., 2002, 36, 299–306 CrossRef CAS PubMed.
- S. Bae and W. Lee, Environ. Sci. Technol., 2014, 48, 2368–2376 CrossRef CAS PubMed.
- G. V. Lowry and K. M. Johnson, Environ. Sci. Technol., 2004, 38, 5208–5216 CrossRef CAS PubMed.
- W. Yan, R. Vasic, A. I. Frenkel and B. E. Koel, Environ. Sci. Technol., 2012, 46, 7018–7026 CrossRef CAS PubMed.
- X.-Q. Li, J. Cao and W.-X. Zhang, Ind. Eng. Chem. Res., 2008, 47, 2131–2139 CrossRef CAS.
- A. Ryu, S.-W. Jeong, A. Jang and H. Choi, Appl. Catal., B, 2011, 105, 128–135 CrossRef CAS.
- G. Sheng, X. Shao, Y. Li, J. Li, H. Dong, W. Cheng, X. Gao and Y. Huang, J. Phys. Chem. A, 2014, 118, 2952–2958 CrossRef CAS PubMed.
- H. Song and E. R. Carraway, Environ. Sci. Technol., 2005, 39, 6237–6245 CrossRef CAS PubMed.
- J. Dong, Y. Zhao, R. Zhao and R. Zhou, J. Environ. Sci., 2010, 22, 1741–1747 CrossRef CAS.
- L. J. Matheson and P. G. Tratnyek, Environ. Sci. Technol., 1994, 28, 2045–2053 CrossRef CAS PubMed.
- J. Klausen, P. J. Vikesland, T. Kohn, D. R. Burris, W. P. Ball and A. L. Roberts, Environ. Sci. Technol., 2003, 37, 1208–1218 CrossRef CAS PubMed.
- X. H. Guan, Y. K. Sun, H. J. Qin, J. X. Li, I. M. C. Lo, D. He and H. R. Dong, Water Res., 2015, 75, 224–248 CrossRef CAS PubMed.
- A. F. Orozco, M. Velimirovic, T. Tosco, A. Kemna, H. Sapion, N. Klaas, R. Sethi and L. Bastiaens, Environ. Sci. Technol., 2015, 49, 5593–5600 CrossRef PubMed.
- E. Dalla Vecchia, M. Luna and R. Sethi, Environ. Sci. Technol., 2009, 43, 8942–8947 CrossRef CAS PubMed.
- S. Comba, D. Dalmazzo, E. Santagata and R. Sethi, J. Hazard. Mater., 2011, 185, 598–605 CrossRef CAS PubMed.
- H. Lee, B. H. Kim, Y. K. Park, S. J. Kim and S. C. Jung, J. Nanomater., 2015, 392537 Search PubMed.
- I. A. Katsoyiannis, T. Ruettimann and S. J. Hug, Environ. Sci. Technol., 2008, 42, 7424–7430 CrossRef CAS PubMed.
- H. S. Lee, H. J. Lee, H. E. Kim, J. Y. Kweon, B. D. Lee and C. H. Lee, J. Hazard. Mater., 2014, 265, 201–207 CrossRef CAS PubMed.
- C. R. Keenan and D. L. Sedlak, Environ. Sci. Technol., 2008, 42, 1262–1267 CrossRef CAS PubMed.
- S. J. Hug and O. Leupin, Environ. Sci. Technol., 2003, 37, 2734–2742 CrossRef CAS PubMed.
- S. H. Joo, A. J. Feitz and T. D. Waite, Environ. Sci. Technol., 2004, 38, 2242–2247 CrossRef CAS PubMed.
- A. J. Feitz, S. H. Joo, J. Guan, Q. Sun, D. L. Sedlak and T. D. Waite, Colloids Surf., A, 2005, 265, 88–94 CrossRef CAS.
- J. Filip, F. Karlický, Z. Marušák, M. Černík, M. Otyepka and R. Zbořil, J. Phys. Chem. C, 2014, 118, 13817–13825 CAS.
- P. Mondal, S. Bhowmick, N. Jullok, W. Y. Ye, W. Van Renterghem, S. Van den Berghe and B. Van der Bruggen, J. Phys. Chem. C, 2014, 118, 21614–21621 CAS.
- X. L. Zhang, B. L. Deng, J. Guo, Y. Wang and Y. Q. Lan, J. Environ. Manage., 2011, 92, 1328–1333 CrossRef CAS PubMed.
- Y. Segura, F. Martinez, J. A. Melero and J. L. G. Fierro, Chem. Eng. J., 2015, 269, 298–305 CrossRef CAS.
- T. X. Liu, X. Li and T. D. Waite, Environ. Sci. Technol., 2013, 47, 7350–7356 CrossRef CAS PubMed.
- T. X. Liu, X. Li and T. D. Waite, Environ. Sci. Technol., 2013, 47, 13712–13720 CrossRef CAS PubMed.
- T. X. Liu, X. Li and T. D. Waite, Environ. Sci. Technol., 2014, 48, 14564–14571 CrossRef CAS PubMed.
- L. Chen, S. Jin, P. H. Fallgren, F. Liu and P. J. S. Colberg, Water Sci. Technol., 2013, 67, 1254–1259 CrossRef CAS PubMed.
- P. V. V. V. Prasad, C. Das and A. K. Golder, Can. J. Chem. Eng., 2011, 89, 1575–1582 CrossRef CAS.
- I. Magario, F. S. García Einschlag, E. H. Rueda, J. Zygadlo and M. L. Ferreira, J. Mol. Catal. A: Chem., 2012, 352, 1–20 CrossRef CAS.
- M. Minella, G. Marchetti, E. De Laurentiis, M. Malandrino, V. Maurino, C. Minero, D. Vione and K. Hanna, Appl. Catal., B, 2014, 154, 102–109 CrossRef.
- P. Avetta, A. Pensato, M. Minella, M. Malandrino, V. Maurino, C. Minero, K. Hanna and D. Vione, Environ. Sci. Technol., 2015, 49, 1043–1050 CrossRef CAS PubMed.
- S. X. Zhang, X. L. Zhao, H. Y. Niu, Y. Shi, Y. Q. Cai and G. B. Jiang, J. Hazard. Mater., 2009, 167, 560–566 CrossRef CAS PubMed.
- F. Tisa, A. A. A. Raman and W. M. A. W. Daud, J. Environ. Manage., 2014, 146, 260–275 CrossRef CAS PubMed.
- S. Gligorovski, R. Strekowski, S. Barbati and D. Vione, Chem. Rev., 2015, 115, 13051–13092 CrossRef CAS PubMed.
- AOAC International, Official Methods of Analysis of AOAC International, Gaithersburg MD, 20th edn, 2016 Search PubMed.
- Y. Ito, Y. Tonogai, H. Suzuki, S. Ogawa, T. Yokoyama, T. Hashizume, H. Santo, K. I. Tanaka, K. Nishigaki and M. Iwaida, J.–Assoc. Off. Anal. Chem., 1981, 64, 1448–1452 CAS.
- R. Cheng, J. L. Wang and W. X. Zhang, Biomed. Environ. Sci., 2007, 20, 410–413 CAS.
- Y. H. Liu, F. L. Yang, P. L. Yue and G. H. Chen, Water Res., 2001, 35, 1887–1890 CrossRef CAS PubMed.
- B. Gunawardana, N. Singhal and P. Swedlund, Environ. Eng. Res., 2011, 16, 187–203 CrossRef.
- H. Daraei and H. Kamali, J. Environ. Anal. Toxicol., 2014, 4, 228 Search PubMed.
- B. Sulzberger and H. Laubscher, Mar. Chem., 1995, 50, 103–115 CrossRef CAS.
- C. Walling, Acc. Chem. Res., 1998, 31, 155–157 CrossRef CAS.
- H. Bataineh, O. Pestovsky and A. Bakac, Chem. Sci., 2012, 3, 1594–1599 RSC.
- J. Gomis, R. F. Vercher, A. M. Amat, D. O. Martire, M. C. Gonzalez, A. B. Prevot, E. Montoneri, A. Arques and L. Carlos, Catal. Today, 2013, 209, 176–180 CrossRef CAS.
- C. Minero, M. Lucchiari, V. Maurino and D. Vione, RSC Adv., 2013, 3, 26443–26450 RSC.
- M. F. Variava, T. L. Church and A. T. Harris, Appl. Catal., B, 2012, 123, 200–207 CrossRef.
- C. C. Kuan, S. Y. Chang and S. L. M. Schroeder, Ind. Eng. Chem. Res., 2015, 54, 8122–8129 CrossRef CAS.
- P. Ghosh, C. Kumar, A. N. Samanta and S. Ray, J. Chem. Technol. Biotechnol., 2012, 87, 914–923 CrossRef CAS.
- C. L. Hsueh, Y. H. Huang, C. C. Wang and C. Y. Chen, Chemosphere, 2005, 58, 1409–1414 CrossRef CAS PubMed.
- U. Bali, E. Catalkaya and F. Sengul, J. Hazard. Mater., 2004, 114, 159–166 CrossRef CAS PubMed.
- M. Vilve, S. Vilhunen, M. Vepsalainen, T. A. Kurniawan, N. Lehtonen, H. Isomaki and M. Sillanpaa, Environ. Sci. Pollut. Res., 2010, 17, 875–884 CrossRef CAS PubMed.
- G. V. Buxton, C. L. Greenstock, W. P. Helman and A. B. Ross, J. Phys. Chem. Ref. Data, 1988, 17, 513–886 CrossRef CAS.
- X. Fang, X. Pan, A. Rahmann, H. P. Schuchmann and C. von Sonntag, Chem.–Eur. J., 1995, 1, 423–429 CrossRef CAS.
- L. Rizzo, Water Res., 2011, 45, 4311–4340 CrossRef CAS PubMed.
- J. A. Zazo, J. A. Casas, C. B. Molina, A. Quintanilla and J. J. Rodriguez, Environ. Sci. Technol., 2007, 41, 7164–7170 CrossRef CAS PubMed.
- S. Harimurti, B. K. Dutta, I. F. B. M. Ariff, S. Chakrabarti and D. Vione, Water, Air, Soil Pollut., 2010, 211, 273–286 CrossRef CAS.
- National Environmental Agency, Allowable limits for trade effluent discharge to sewer/watercourse/controlled watercourse, http://www.nea.gov.sg, last accessed May 2016.
- D. He, J. Ma, R. N. Collins and T. D. Waite, Environ. Sci. Technol., 2016, 50, 3820–3828 CrossRef CAS PubMed.
- Y. Liu and G. V. Lowry, Environ. Sci. Technol., 2006, 40, 6085–6090 CrossRef CAS PubMed.
- S. Bae and K. Hanna, Environ. Sci. Technol., 2015, 49, 10536–10543 CrossRef CAS PubMed.
- X. Q. Li and W. X. Zhang, Langmuir, 2006, 22, 4638–4642 CrossRef CAS PubMed.
- H. Luo, S. Jin, P. H. Fallgren, P. J. S. Colberg and P. A. Johnson, Chem. Eng. J., 2010, 160, 185–189 CrossRef CAS.
- S. Bae, S. Gim, H. Kim and K. Hanna, Appl. Catal., B, 2016, 182, 541–549 CrossRef CAS.
- M. F. Hochella, S. K. Lower, P. A. Maurice, R. L. Penn, N. Sahai and D. L. Sparks, Science, 2008, 319, 1631–1635 CrossRef CAS PubMed.
- R. M. Cornell and U. Schwertmann, The Iron Oxides: Structure, Properties, Reactions, Occurrences and Uses, John Wiley & Sons, Weinheim, 2006 Search PubMed.
- R. Matta, K. Hanna and S. Chiron, Sci. Total Environ., 2007, 385, 242–251 CrossRef CAS PubMed.
- X. Xue, K. Hanna and N. Deng, J. Hazard. Mater., 2009, 166, 407–414 CrossRef CAS PubMed.
- G. Hurwitz, P. Pornwongthong, S. Mahendra and E. M. V. Hoek, Chem. Eng. J., 2014, 240, 235–243 CrossRef CAS.
- P. Nissenson, D. Dabdub, R. Das, V. Maurino, C. Minero and D. Vione, Atmos. Environ., 2010, 44, 4859–4866 CrossRef CAS.
- V. Sarathy, P. G. Tratnyek, J. T. Nurmi, D. R. Baer, J. E. Amonette, C. Chun, R. L. Penn and E. J. Reardon, J. Phys. Chem. C, 2008, 112, 2286–2293 CAS.
- L. Demarchis, M. Minella, R. Nisticò, V. Maurino, C. Minero and D. Vione, J. Photochem. Photobiol., A, 2015, 307, 99–107 CrossRef.
- Y. L. Wu, M. Passananti, M. Brigante, W. B. Dong and G. Mailhot, Environ. Sci. Pollut. Res., 2014, 21, 12154–12162 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra17515e |
| This journal is © The Royal Society of Chemistry 2016 |