
An optical fibre sensor for remotely detecting water traces in organic solvents
Mengke
Han,  ab
Youhong
Tang
g
and
ab
Youhong
Tang
g
and
ab
Youhong
Tang
g
and
Abstract
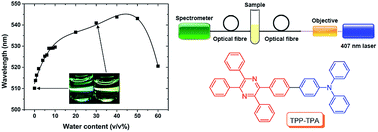
Tetraphenylpyrazine-triphenylamine (TPP-TPA) was used to detect water traces in organic solvents by monitoring the shift of the fluorescence peak wavelength. This wavelength based method avoids the intrinsic problems of fluorescence intensity change based methods. The use of optical fibres for the detection provides a remote and field-deployable sensing ability.
 Please wait while we load your content...
Please wait while we load your content...

