
Promotion mechanism of pyridine N-doped carbocatalyst for SO2 oxidation†
Jieyuan Lia,
Jie Liub,
Shi Yina,
Yongjun Liuad,
Jianjun Liad,
Wanglai Cen*cd and
Yinghao Chu*ad
aCollege of Architecture and Environment, Sichuan University, P. R. China. E-mail: chuyinghao@scu.edu.cn
bResources and Environmental College, Chengdu University of Information Technology, P. R. China
cInstitute of New Energy and Low Carbon Technology, Sichuan University, P. R. China. E-mail: cenwanglai@163.com
dNational Engineering Research Center for Flue Gas Desulfurization, P. R. China
First published on 29th August 2016
Abstract
Pyridine N (PyN) doped carbon materials have long been recognized as promising catalysts for SO2 oxidation under mild conditions, but our understanding of what is happening at the atomic level is still limited. Herein, the local structure and promotion mechanism of PyN in carbon materials for the catalytic oxidation of SO2 were investigated on graphene model catalysts by using density functional theory. A type of defect involving three PyN atoms around a single C atom vacancy was found to be active for both the dissociation of O2 and the further oxidation of SO2. It is worth mentioning that both PyN and the adjacent C atoms are primary active sites. Additionally, a switch effect of pyridine N-oxide was identified, which can suppress or enhance the oxidation capacity of surface oxygen species for SO2 oxidation. These results provide a mechanistic explanation for the low temperature catalytic oxidation of SO2 by PyN-doped carbon materials and offer insight for the design of new carbon-based catalysts.
1. Introduction
The development of cost efficient technology for the emission control of SO2 derived from fossil fuel burning processes is still required for environmental protection. Carbon materials have long been recognized as promising candidates for the catalytic oxidation and recovery of sulfuric acid or SO2 at mild conditions (1 atm and 50–100 °C).1,2 However, the mechanism of the catalytic reaction at the atomic level, particularly involving the local structure and the role of N heteroatoms doped into the host carbon materials, is still unclear, which hinders the advancement of carbocatalysts.3,4Recently, doping by heteroatoms has extensively been explored to enhance the catalytic activity of sluggish raw carbon materials, especially graphene (GP) as a model catalyst.5,6 Among them, nitrogen has been found to be a promising candidate for both the oxygen reduction reaction (ORR) in electrocatalysis7–9 and the oxidation reaction in thermocatalysis.10,11 Our theoretical investigation was motivated by the unique promotion effects of pyridine N (PyN) in carbon materials for SO2 catalytic oxidation.12,13
It is conventionally recognized that the SO2 oxidation reaction (SOR) follows the Langmuir–Hinshelwood mechanism.12 Molecular oxygen is adsorbed and then dissociates to form surface active oxygen species, which subsequently oxidizes the adsorbed SO2. Both experimental14 and theoretical works15 have shown that the SO2 molecule can be readily oxidized by epoxy groups on a pristine GP surface. However, the barrier for the O2 dissociation reaction (ODR) is likely too high to be overcome.11,16 The barrier for the ODR can be significantly reduced by S/O doping, but this causes the corresponding SOR barrier to be increased to at least 0.8 eV.16 These results indicate that there should be a compromise between the ODR and SOR when doping with hereroatoms.
It has been experimentally found that the catalytic oxidation of SO2 by O2 with carbon materials can be promoted by N-doping,13,17 predominantly in the form of PyN,13 implying that the compromise between the ODR and SOR on a carbocatalyst could be reconciled through the use of nitrogen doping. However, the mechanism of such a balance is still under controversy. The debate mainly focuses on whether the activity promotion of the carbocatalyst is due to PyN or graphitic N (GrN).7,18 According to Yang,11 the introduction of GrN can reduce the barrier of the ODR to 0.2 eV. This can also result in the super catalytic capacity for the oxidation of the C–H bond, by changing the electronic structure of the adjacent carbon atoms and stimulating their chemical reactivity.19 However, the electrochemical ORR performance of PyN has been found to be preferable to that of GrN, and it is explained in a similar way.7,20,21 This controversy may be due to the differences between the mechanisms for the electro- and thermo-catalysis of carbocatalysts, which has seldom been addressed. Additionally, the role of the doped nitrogen atoms might be underestimated, since they are Lewis basic centers that donate electrons to oxidants for catalytic oxidation. Hence, it is still imperative to identify the doping pattern and promotion mechanism of nitrogen-doped carbon materials for SO2 catalytic oxidation.
The edge sites of GP or its derivatives have been frequently investigated for PyN-doping.9,21,22 Vacancies in the GP plane, with single or double carbon atoms removed, are commonly observed,23–25 and can be useful for nitrogen doping to obtain a higher PyN concentration. Consequently, in this context, two kinds of vacancies will be considered for PyN-doping, with various dopant concentrations, to investigate the possible mechanism for SO2 catalytic oxidation by molecular O2. The subtle role of the doped nitrogen, in addition to that of the adjacent carbon atoms, will also be addressed.
2. Methods and models
2.1 Computational models
A supercell of GP including 72 C atoms was employed as a model catalyst. The lattice parameters were relaxed to 12.78 × 14.76 Å2 (Fig. 1a), with a vacuum region of 20.00 Å to minimize the interaction between different layers.26 Single or double C atoms were removed to fabricate vacancies contained in GP (denoted as VG or DVG respectively, as shown in Fig. 1b and c). There are three elementary forms of DVG, which are generated by the coalescence of two VGs (555-777 and 555-6-777) or by removing two neighboring atoms (5-8-5), among which the 5-8-5 configuration has been reported to be of high activity.27 Consequently, the VG and DVG (5-8-5) configurations are used as our models for N substitution.23,24
supercell of GP including 72 C atoms was employed as a model catalyst. The lattice parameters were relaxed to 12.78 × 14.76 Å2 (Fig. 1a), with a vacuum region of 20.00 Å to minimize the interaction between different layers.26 Single or double C atoms were removed to fabricate vacancies contained in GP (denoted as VG or DVG respectively, as shown in Fig. 1b and c). There are three elementary forms of DVG, which are generated by the coalescence of two VGs (555-777 and 555-6-777) or by removing two neighboring atoms (5-8-5), among which the 5-8-5 configuration has been reported to be of high activity.27 Consequently, the VG and DVG (5-8-5) configurations are used as our models for N substitution.23,24
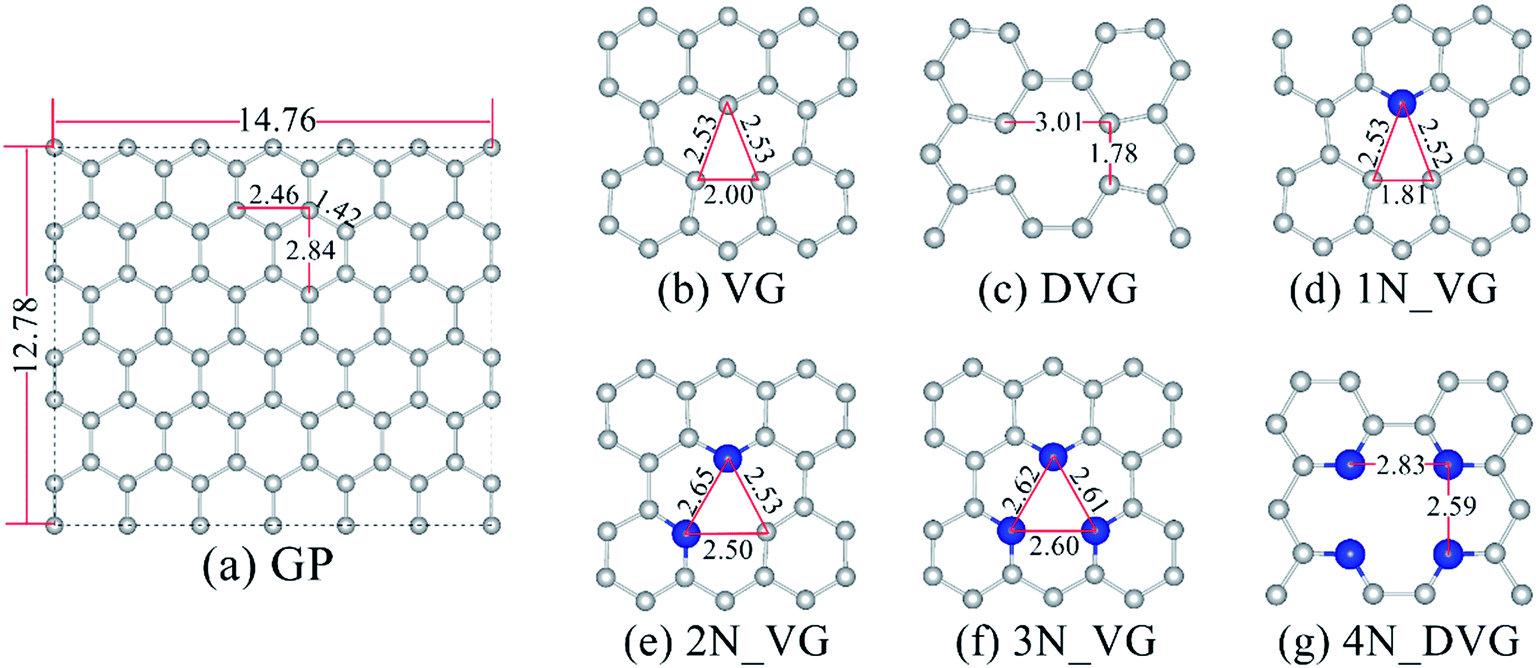 | ||
| Fig. 1 Relaxed structures of perfect (a), vacancy defected (b and c) and N doped (d–g) graphene. The gray (blue) ball stands for C (N) atoms. All lengths are given in Å. | ||
For the doping of N atoms into VG or DVG, the N atoms are inclined to substitute the unsaturated C atoms at the boundary of the vacancies in the GP plane.19 Our test calculations confirm this trend (Fig. S1†). When 1–3 unsaturated C atoms are substituted by N atoms, the resulting configurations are denoted as 1–3N_VG (Fig. 1d–f). 3N_VG has been regarded as the Czerw model.28 Accordingly, the four PyN-doped DVG configuration is denoted as 4N_DVG (Fig. 1g).
2.2 Computational methods
All the spin polarized DFT-D2 calculations were carried out by employing the VASP5.3 code,29,30 using the generalized gradient approximation with the Perdew–Burke–Ernzerhof (PBE) exchange and correlation functional.31 A plane-wave basis set with a cut-off energy of 400 eV within the framework of the projector-augmented wave (PAW) method32,33 was employed. The Gaussian smearing width was set to be 0.2 eV. The van der Waals (vdW) correction is described by the Grimme method34 with default parameters. The Brillouin zone was sampled using a 3 × 3 × 1 k-points mesh. All atoms, except those on the boundary, were allowed to relax and converge to 0.02 eV Å−1 for all systems. The calculated bond lengths are consistent with published values.35,36 The nudged elastic band (NEB) method37,38 was used to search the minimum energy pathways (MEP) from an initial state (IS) to its final state (FS), and the transitional state (TS) was localized with the climbing image method and verified with a single imaginary frequency.The adsorption energy (ΔEads) is defined as
| ΔEads = Etot − (Emol + Eslab), |
The total doping energy (Ed)29 is defined as
| Ed = Edoped − (Eun-doped − EC × N + EN × N), |
The substep doping energy (Es) is defined as the extra energy cost for every other N atom doped on the substrate. The reference energy for C is the energy of a C atom in pristine graphene, and that for N is the energy of an N atom in an isolated N2 molecule.
3. Results
3.1 PyN-doped structures and adsorption of O2
The doping energy results (Table S1†) indicate that all the unsaturated C atoms are thermodynamically to be substituted by nitrogen atoms. As shown in Fig. 1b, the three unsaturated C atoms in VG are arranged in an isosceles triangle. When the first N atom is introduced to form 1N_VG, the base is shortened remarkably from 2.00 Å to 1.81 Å, which may lead to a strong covalent interaction between the two unsaturated C atoms. The substep doping energy for 1N_VG is −2.06 eV (Table S1†), while that for 2N_VG is reduced to −0.89 eV. The reduction can be ascribed to the breaking of the covalent interaction between the two unsaturated C atoms in VG. The low substep doping energy for 1N_DVG (−0.09 eV) and 3N_DVG (−0.77 eV) can be explained in the same way.Table 1 summarizes the calculated results for O2 adsorption on different PyN-doped surfaces. Corresponding adsorption configurations can be found in Fig. S2.† The negative adsorption energy (ΔEads) implies that all the adsorptions are thermodynamically preferable. The adsorption energy for O2/1N_VG is as high as −1.64 eV, and that for O2/2N_VG is even higher. The C–O distance (dC–O) is 1.46 Å for O2/1N_VG and 1.32 Å for O2/2N_VG. The Bader charge is −1.09 e and −1.22 e, respectively. All these results indicate the strong chemisorption of the O2 molecule at unsaturated C atom(s). Subsequently, the adsorbed O2 becomes remarkably activated as the O–O bond is extended to 1.46 Å on 1N_VG, in comparison with the value of 1.26 Å for the free O2 molecule. For O2/2N_VG, the O–O bond is activated to 1.35 Å. However, the adsorbed O2 can become dissociated to form carbonyl groups, which are inactive for SO2 oxidation according to our previous work16 and will be addressed in Section 3.2.
| Conf. | ΔEads, eV | dC–O/N–O, Å | dO–O, Å | Δq, e |
|---|---|---|---|---|
| a dC–O/N–O: distance between O and the closest C/N; dO–O: length of O–O in O2; Δq: total charge of the absorbed O2 using the Bader method,39 where a negative value means that electrons are transferred to O2. | ||||
| O2/1N_VG | −1.64 | 1.46/— | 1.46 | −1.09 |
| O2/2N_VG | −2.07 | 1.32/— | 1.35 | −1.22 |
| O2/3N_VG | −0.33 | 2.00/1.98 | 1.28 | −0.19 |
| O2/4N_DVG | −0.22 | —/2.98 | 1.25 | −0.10 |
The adsorption energy for O2 on 3N_VG is much lower than that on 1N_VG and 2N_VG, with a value of −0.33 eV. The adsorption distance is 2.00 Å for dC–O, and 1.98 Å for dN–O. These results indicate that the adsorption of O2 on PyN saturated structures is a weak and physical adsorption, which is consistent with the bond length and Bader charge results of the adsorbed O2 molecule. The same situation is also observed for O2 adsorption on 4N_VG, as listed in Table 1.
As shown in the charge difference plots in Fig. 2b and d, both PyN and adjacent C atoms are involved in the adsorption of O2 molecules. Charge density and Bader population analysis (Fig. 2a and c) show that PyN sites are Lewis base centers where electrons concentrate, while the C atoms next to them are positively charged. On the approach of O2 during the adsorption, electrons are transferred from N1 to O2 through the O atom closest to it. Then, the other O atom of the O2 molecule is repelled by N2 and moves to the top site of the C atom beside N2.
 | ||
| Fig. 2 Charge density (a and c) and charge difference (b and d) analysis. Bader charges are labeled on selected atoms. Charge accumulation is in blue and depletion in yellow for (b) and (d). Isosurfaces: 0.25 eV Å−3 for (a) and (c), 0.05 eV Å−3 for (b) and 0.01 eV Å−3 for (d). | ||
The adsorption of O2 on 3N_VG is energetically preferred to its absorption on 4N_DVG by 0.11 eV, and the negatively charged O–O bond of the former is longer than that of the latter, implying the activity for the dissociation of O2 on 3N_VG might be higher than that on 4N_DVG. Furthermore, we believe that the unique electronic structure of 3N_VG is important to balance the dissociation of O2 and the oxidation of SO2, which will be addressed in the next section and in the Discussion.
3.2 O2 dissociation and SO2 oxidation
 | ||
| Fig. 3 Catalytic oxidation of SO2 on 1N_VG: (a) ODR1, (b) SOR1, (c) SOR2 and (d) ODR2. Blue, red, yellow and grey spheres represent N, O, S and C atoms, respectively. All the lengths and energies are given in Å and eV respectively. | ||
The two carbonyl groups must be consumed by the oxidation of two SO2 molecules for a complete catalytic reaction loop. The barrier for the first oxidation by one carbonyl group is 0.12 eV (Fig. 3b). The left carbonyl group sinks into the GP plane to form an ether group. The ether group is extremely inactive for the oxidation of a second SO2 molecule as the barrier is as high as 4.07 eV (Fig. 3c), with a net energy cost of 2.39 eV. The substrate structure is also inactive for ODR2 (Fig. 3d), as the barrier is as high as 2.62 eV. Hence, the 1N_VG should be inactive for the catalytic oxidation of SO2, leading to an N/O co-doped configuration.
For 2N_VG, a carbonyl group and an epoxy group are formed during the ODR1 (Fig. S4†). The barrier is 0.35 eV (Fig. S5a†). The situation is basically the same as that for 1N_VG. The epoxy group is supposed to be active for oxidation,15 but the calculated barrier is as high as 0.82 eV for SOR1 (Fig. S5b†). The barrier for SOR2 is 2.19 eV, with a net energy cost of 1.54 eV (Fig. S5c†).
Consequently, both 1N_VG and 2N_VG are inactive for the catalytic oxidation of SO2. The main obstacle is the low activity of surface oxygen species derived from the dissociation of O2, particularly the ether group for 1N_VG and the carbonyl group for 2N_VG. The formation of these two kinds of sluggish oxygen species is due to unsaturated C atoms. Hence, the catalytic activity might be enhanced by substituting all unsaturated C atoms with PyN.
According to our test calculations (Fig. S6†), O2 tends to dissociate to form an epoxy group and a pyridine N-oxide (N–O) (Fig. 4a). The dissociation barrier is 0.68 eV, with 0.64 eV released. However, a barrier as high as 2.34 eV has been reported, with a 1.2 eV cost.11 The divergence may be due to the fact that a wrong configuration with two epoxy groups is used as the final product of the ODR in the reference. Compared to 1N_VG and 2N_VG, the dissociation barrier is slightly higher, but it is much lower than 1.78 eV for the pristine GP surface.16
 | ||
| Fig. 4 Catalytic oxidation of SO2 on 3N_VG (path I): (a) ODR, (b) SOR1, (c) SOR2. The color coding is the same as in Fig. 3. All the lengths and energies are given in Å and eV, respectively. | ||
Due to the different order for the consumption of N–O and epoxy, there might be two reaction pathways. In the first pathway (Fig. 4), the epoxy group is first used for the SOR1. The oxidation barrier is 0.75 eV. Then the second oxidation barrier of N–O is 0.40 eV. It seems that N–O is more active for oxidation than the epoxy group, which is in contrast to our previous findings.15
In path II, the first oxidation barrier of N–O is 0.48 eV (Fig. 5a). Then the succeeding barrier by epoxy is 0.47 eV (Fig. 5b). Both of the two oxidations are thermodynamically feasible. The oxidation of SO2 in path II (Fig. 5) is much smoother than that of path I (Fig. 4), because the main barrier (0.48 eV) of the latter is smaller than that of the former (0.75 eV).
 | ||
| Fig. 5 Catalytic oxidation of SO2 on 3N_VG (path II): (a) SOR1 and (b) SOR2. The color coding is the same as in Fig. 3. All the lengths and energies are given in Å and eV, respectively. | ||
In summary, for the catalytic oxidation of SO2 on the three kinds of PyN-doped VG, the rate-determining barrier decreases in the order 1N_VG (4.07 eV) > 2N_VG (2.19 eV) > 3N_VG (0.68 eV). Saturated PyN-doped 3N_VG is found to reduce the ODR barrier tremendously but maintain the SOR barrier at a low value.
For comparison, saturated PyN-doped 4N_DVG (Fig. 6) was employed for SO2 catalytic oxidation. The dissociation of O2 is inclined to form two N–O groups (Fig. S7†). The barrier is 1.25 eV (Fig. 6a), which is higher than that of 3N_VG (0.68 eV). For the SOR1, due to one of the N–O groups, the barrier is 0.58 eV. Then the barrier is increased to 0.84 eV for the SOR2. Both oxidation barriers are higher than that of 3N_VG. Hence, 4N_DVG is less active than 3N_VG for SO2 catalytic oxidation.
 | ||
| Fig. 6 Catalytic oxidation of SO2 on 4N_DVG: (a) ODR, (b) SOR1 and (c) SOR2. The color coding is the same as in Fig. 3. All the lengths and energies are given in Å and eV, respectively. Negative values mean that heat is released. | ||
4. Discussion
The highest activity for SO2 catalytic oxidation was found using 3N_VG. The doped structure reaches an excellent compromise between the energy barriers of the ODR and the subsequent SOR. We believe that this kind of balance is due to two interesting phenomena, which have not been reported before: (1) both PyN and the adjacent C atoms are involved in the activation of adsorbed O2 molecules for their dissociation, and (2) N–O is of high activity for the SOR. Detailed electronic structure analysis may help in determining the underlying principles.Generally, for an N-doped GP surface, the N sites are the strongest Lewis base centers for O2 adsorption, since N atoms possess much stronger electronegativity than C atoms. However, for O2/1N_VG with unsaturated N-doping, the adsorbed O atoms are located between two unsaturated C atoms as shown in the configuration. For O2/3N_VG (Fig. 2b and c) and O2/4N_VG (Fig. 2e and f), the O atoms are located close to an N atom at one end and a C atom at the other. These kinds of O2 adsorption configurations are consistent with typical ORR reports,7 which deduce that the adjacent C atoms (to the doped N atoms) are Lewis basic and take the role of active centers. In this view, however, the Lewis basicity of the N atom is over neglected.
Fig. 7a presents the selected projected density of states (PDOS) of C 2p and N 2p for 1N_VG before O2 adsorption. This figure demonstrates that N 2p electrons are primarily localized in the plane (px + py) of GP. It is worth noting that the occupied N 2pz near the Fermi level (−2 to 0 eV) is lower than C 2pz, which may play a key role in the donation of electrons from the 1N_VG surface to the O2 molecule, rather than the total electronegativity or Lewis basicity of the surface atoms.
 | ||
| Fig. 7 Projected density of states (PDOS) for different N-doped GP surfaces: (a) 1N_VG, (b) 3N_VG and (c) 4N_DVG. The selected N and C atoms have been labeled in the corresponding insets. The Fermi level is set to 0 eV. | ||
On the contrary, for the situations of 3N_VG (Fig. 7b) and 4N_DVG (Fig. 7c), the occupied N 2pz near the Fermi level is higher than C 2pz, leading to the preferable adsorption of O2 to one of the N atoms at one end. During the adsorption to the N atom, the O2 molecule is negatively charged and is inclined to be adsorbed to a positively charged C atom at the other end through the Coulomb interaction, which is consistent with the relaxed O2 adsorption configurations as mentioned before. Compared to N 2pz of 4N_DVG, the N 2pz from −1 eV to the Fermi level of 3N_VG is higher, which results in more electrons being transferred to the adsorbed O2 molecule and the lowering of the barrier for its dissociation. Since the number of electrons obtained by oxygen from 4N_DVG is less than that of 3N_VG (Table 1), the result that both dissociated O atoms are situated on negatively charged PyN of 4N_DVG is conceivable.
For the recovery of catalytic activity in a complete catalytic loop, both of the oxygen species derived from the O2 dissociation are consumed by the oxidation of two SO2 molecules. We know that N–O is more active than epoxy in 2O/3N_VG, and it is of interest to find that the N–O species plays a “switch” role on the catalytic activity of N-doped GP. Its consumption reduces the barrier of the left epoxy group from 0.75 eV to 0.47 eV on 3N_VG, while raising the barrier of the second N–O species from 0.58 eV to 0.84 eV on 4N_DVG.
The switch effects can be addressed well by employing PDOS and electron localization function (ELF) analysis as shown in Fig. 8. For 3N_VG, the covalent peak α between the epoxy group and the carbon atoms connected to it is weakened slightly and the localized O 2p peak β just below the Fermi level is enhanced and shifted to a higher energy level by 0.18 eV (Fig. 8a and b), due to the consumption of N–O. In contrast, for 4N_DVG, the N–O covalent peak α is enhanced and the O 2p peak γ just below the Fermi level is shrunken and merges with the next peak β (Fig. 8c and d). ELFs plots in Fig. 8a′–d′ additionally confirm that the covalent bonding interaction is weakened for epoxy in 3N_VG and enhanced for N–O in 4N_DVG.
 | ||
| Fig. 8 Projected density of states (a–d) and electron localization function (a′–d′) for different oxygen-containing groups (circled in the corresponding insets) on different N-doped GP surfaces. (a) Epoxy in 2O/3N_VG, (b) epoxy in 1O/3N_VG, (c) N–O in 2O/4N_DVG and (d) N–O in 1O/4N_DVG. (a′–d′) are in the same order. Blue, red and grey spheres represent N, O and C atoms, respectively. | ||
Finally, it is found that the SOR activity of the three main different oxygen species for the first SOR decreases in the following order: N–O in 2O/3N_VG > N–O in 2O/4N_DVG > epoxy in 2O/3N_VG. This trend can be ascribed to the energy levels of the highest occupied molecular orbitals (HOMOs) as shown in Fig. 9. The HOMO peak center of N–O in 2O/4N_DVG is 1.13 eV under the Fermi level, which is higher than that of epoxy in 2O/3N_VG by 1.35 eV. The HOMO peak of N–O in 2O/3N_VG is located at −0.85 eV, quite close to the Fermi level, indicating that the N–O bond in 2O/3N_VG can be broken more easily than the other two cases. Therefore, the 3N_VG configuration possesses the highest activity for the catalytic oxidation of SO2.
 | ||
| Fig. 9 PDOS for different surface oxygen species (circled in the corresponding insets) for the oxidation of SO2: (a) N–O in 2O/3N_VG, (b) N–O in 2O/4N_DVG, and (c) epoxy in 2O/3N_VG. The color coding is the same as in Fig. 8. | ||
5. Conclusion
Complete catalytic reaction loops for the oxidation of SO2 by O2 on several different pyridine N-doped graphene materials were investigated by using DFT-D2 calculations. A 3N_VG model with three pyridine N atoms substituting all the three C atoms around a single C atom vacancy is found to be the optimized doping configuration, and is active for both the dissociation of O2 and the two following oxidation steps of SO2. The barrier for O2 dissociation is 0.68 eV, which involves the formation of a pyridine N-oxide (N–O) and an epoxy group. After this, both the pyridine N-oxide and the epoxy group are active for SO2 oxidation, with barriers of 0.48 and 0.47 eV, respectively. These results provide a possible local structure of pyridine N-doped carbon-based materials for the catalytic oxidation of SO2.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (51508356, 51378325) and Science and Technology Support Program of Sichuan Province (2014GZ0213). We also acknowledge the National Supercomputer Center in Shenzhen of China and the Institute of New Energy and Low Carbon Technology of Sichuan University for computational support.References
- M. Lzquierdo, B. Rudio and C. Mayoral, Fuel, 2003, 82, 147–151 CrossRef.
- U. M. Azhar, Y. Toru, R. Ochiai and E. Sasaoka, Energy Fuels, 2008, 22, 2284–2289 CrossRef.
- S. Navalon, A. Dhakshinamoorthy, M. Alvaro and H. Garcia, Chem. Rev., 2014, 114, 6179–6191 CrossRef CAS PubMed.
- D. R. Dreyer, H. P. Jia and C. W. Bielawski, Angew. Chem., Int. Ed., 2010, 49, 6813–6816 CAS.
- C. L. Su and K. P. Loh, Acc. Chem. Res., 2012, 46, 2275–2285 CrossRef PubMed.
- C. Huang, C. Li and G. Shi, Energy Environ. Sci., 2012, 5, 8848–8868 CAS.
- D. Guo, R. Shibuya, C. Akiba, S. Saji, T. Kondo and J. Nakamura, Science, 2016, 351, 361–365 CrossRef CAS PubMed.
- F. Gao, G.-L. Zhao and S. Yang, ACS Catal., 2014, 4, 1267–1273 CrossRef CAS.
- M. Li, L. Zhang, Q. Xu, J. Niu and Z. Xia, J. Catal., 2014, 314, 66–72 CrossRef CAS.
- W. Li, Y. Gao, W. Chen, P. Tang, W. Li, Z. Shi, D. Su, J. Wang and D. Ma, ACS Catal., 2014, 4, 1261–1266 CrossRef CAS.
- S. Ni, Z. Li and J. Yang, Nanoscale, 2012, 4, 1184–1189 RSC.
- A. A. Lizzio and J. A. DeBarr, Energy Fuels, 1997, 11, 284–291 CrossRef CAS.
- E. Raymundo-Piñero, D. Cazorla-Amorós and A. Linares-Solano, Carbon, 2003, 41, 1925–1932 CrossRef.
- Y. Long, C. Zhang, X. Wang, J. Gao, W. Wang and Y. Liu, J. Mater. Chem., 2011, 21, 13934–13941 RSC.
- W. Cen, M. Hou, J. Liu, S. Yuan, Y. Liu and Y. Chu, RSC Adv., 2015, 5, 22802–22810 RSC.
- M. Hou, W. Cen, F. Nan, J. Li, Y. Chu and H. Yin, RSC Adv., 2016, 6, 7015–7021 RSC.
- A. Peñas-Sanjuán, R. López-Garzón, M. Domingo-García, F. J. López-Garzón, M. Melguizo and M. Pérez-Mendoza, Carbon, 2012, 50, 3977–3986 CrossRef.
- T. Kondo, S. Casolo, T. Suzuki, T. Shikano, M. Sakurai, Y. Harada, M. Saito, M. Oshima, M. I. Trioni, G. F. Tantardini and J. Nakamura, Physical Review B: Condensed Matter and Materials Physics, 2012, 86, 035436 CrossRef.
- Y. Gao, G. Hu, J. Zhong, Z. Shi, Y. Zhu, D. S. Su, J. Wang, X. Bao and D. Ma, Angew. Chem., Int. Ed., 2013, 52, 2109–2113 CrossRef CAS PubMed.
- L. Zhang, J. Niu, L. Dai and Z. Xia, Langmuir, 2012, 28, 7542–7550 CrossRef CAS PubMed.
- L. Zhang and Z. Xia, J. Phys. Chem. C, 2011, 115, 11170–11176 CAS.
- H. Kim, K. Lee, S. I. Woo and Y. Jung, Phys. Chem. Chem. Phys., 2011, 13, 17505–17510 RSC.
- K. S. Hashimoto, A. Gloter, K. Urita and S. Iijima, Nature, 2004, 430, 870–873 CrossRef PubMed.
- F. Banhart and J. Kotakoski, ACS Nano, 2011, 5, 26–41 CrossRef CAS PubMed.
- F. Ducastelle, Physical Review B: Condensed Matter and Materials Physics, 2013, 88, 075413 CrossRef.
- S. Tang and Z. Cao, J. Chem. Phys., 2011, 134, 044710 CrossRef PubMed.
- H. Zhao, C. Sun, Z. Jin, D.-W. Wang, X. Yan, Z. Chen, G. Zhu and X. Yao, J. Mater. Chem. A, 2015, 3, 11736–11739 CAS.
- R. Czerw and B. Foley, Nano Lett., 2001, 1, 457–460 CrossRef CAS.
- G. Kresse and J. Furthmuller, Physical Review B: Condensed Matter and Materials Physics, 1996, 54, 11169–11186 CrossRef CAS.
- G. Kresse and J. Furthmuller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- P. E. Blochl, Physical Review B: Condensed Matter and Materials Physics, 1994, 50, 17953–17979 CrossRef.
- G. Kresse and D. Joubert, Physical Review B: Condensed Matter and Materials Physics, 1999, 59, 1758–1775 CrossRef CAS.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- E. Fuente, J. A. Menendez, M. A. Diez, D. Suarez and M. A. Montes-Moran, J. Chem. Phys., 2003, 107, 6350–6359 CrossRef CAS.
- D. R. Lide, Handbook of chemistry and physics, CRC Press, 2003 Search PubMed.
- G. Henkelman and H. Jónsson, J. Chem. Phys., 2000, 113, 9978 CrossRef CAS.
- G. Henkelman, B. P. Uberuaga and H. Jónsson, J. Chem. Phys., 2000, 113, 9901 CrossRef CAS.
- R. Bader, Atoms in Molecules: A Quantum Theory, Oxford University Press, 1994 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra17349g |
| This journal is © The Royal Society of Chemistry 2016 |