
Encapsulation of multi-stimuli AIE active platinum(II) complex: a facile and dry approach for luminescent mesoporous silica†
Sheik Saleem Pasha,
Parvej Alam,
Amrit Sarmah,
Ram Kinkar Roy and
Inamur Rahaman Laskar*
Department of Chemistry, Birla Institute of Technology and Science, Pilani Campus, Pilani, Rajasthan, India. E-mail: ir_laskar@bits-pilani.ac.in
First published on 17th August 2016
Abstract
Luminescent materials have great potential in diverse applications in their solid state. Because these materials are subject to the aggregation-caused quenching (ACQ) effect, increasing attention is focused on synthesizing aggregation-induced emission (AIE) active materials to avoid the ACQ effect. Herein a new class of AIE active, excimeric platinum(II) complex, [Pt(C^N)(L1)(Cl)], 3 [C^N = 2-phenylpyridine; L1 = N1-tritylethane-1,2-diamine] is reported. The complex 3 exhibited mechanofluorochromism (MFC) and thereby transformed into an orange-emitting complex, 3a, upon grinding. Crushing of 3 (or 3a) with meso-structured silica produced a luminescent composite material, 3b, and thereby the AIE Pt(II) complex moved into the mesopores and the process signaled with a drastic change of emission color (yellow → green). The solid-state luminescent behaviour of these complexes was thoroughly studied. The photophysical properties were also supported by TD-DFT based theoretical study.
In the past few decades, there has been enormous interest in platinum(II) based phosphorescent complexes due to their rich spectroscopic and luminescence properties such as high luminescence quantum yields, long emission lifetimes, large Stoke shifts, excellent emission properties, easy color tunability and high photo-stability as compared to the traditional fluorescent dyes.1 These intriguing properties make the platinum(II) complexes potentially useful in many applications such as chemo-sensors, non-linear optical (NLO) materials, photo catalysts, bio-imaging, optical power limiting materials (OPL) and organic light-emitting devices (OLEDs), and drug discovery, among others.2 However, the square planar structure of cyclometalated Pt(II) complexes often forms excimers (or exciplexes) because of Pt–Pt or π–π interactions upon excitation.3 These interactions generate a new metal–metal-to-ligand charge transfer (MMLCT) excited state that results in broad and red-shifted emissions in comparison to monomeric emissions.4 The Pt–Pt and π–π interactions are very sensitive to external stimuli and hence draw attention in promising applications such as optical recording, memory sensing and display devices.5
The emission intensity of the luminescent materials is in general quenched in concentrated solutions or in aggregated states, known as the aggregation-caused quenching (ACQ) effect. This ACQ effect hinders the proper usage of luminescent materials in many applications.6 In 2001, Tang and co-workers7 synthesized a silole derivative that was almost non-emissive in solution state but strongly emissive in aggregated state. This was an utterly opposite phenomenon of ACQ renowned as aggregation-induced emission (AIE), an effective approach to achieve maximum emission intensity in the aggregated/solid state.7 In the last decade, it has been observed that development of AIE luminophores are mainly limited to organic luminophores, but the AIE active metal complexes, and especially Pt(II) complexes, had not been adequately explored.8
The luminescent Pt(II) complexes have astounding records in the field of bio-imaging. Unfortunately, background fluorescence scattered light and water solubility of such complexes pose great obstacles in these applications. Development of biocompatible, mesoporous silica-encapsulated AIE material could be a better alternative to tackle such problems.9 Impregnation of luminescent materials into the mesoporous silica has received great interest because they result in high thermal and photo stabilities, bio-compatibility, large accessible pore sizes and periodic nano-scale pore spacing. In general, methodologies adopted to incorporate luminescent materials into silica pores were mainly wet-based techniques.10
Recently several research groups have found this rare AIE observation in square planar Pt(II) complexes.8 The design, syntheses and investigation of mechanistic pathways of AIE Pt(II) complexes comprise an immense and exigent task for organometallic-chemists.8 Our group has been extensively working on the design and syntheses of new AIE active Ir(III) and Pt(II)-based complexes.11,8c
After successful syntheses of several Ir(III)-based AIE active complexes, we obtained a breakthrough in syntheses of two new AIE active Pt(II) complexes with rotary groups, and the restricted intramolecular rotation (RIR) was established as the mechanism for AIE.8c In continuing our previous work, we have designed and synthesized a new monocyclometalated Pt(II) complex by linking a bulky rotating unit (trityl-) in the non-coordinating site of ethane-1,2-diamine that generated an AIE active molecule. This molecule was successfully incorporated into the pores of mesoporous silica in a simple dry technique with a concomitant sharp change of emission color. Additionally, this AIE active Pt(II) molecule exhibited mechanofluorochromic properties. The causes of such luminescent property changes were explored. We performed computational calculations of 3, which were correlated to the measured spectroscopic data.
Complex 3 was synthesized in a convenient route by using [Pt(C^N)(CH^N)Cl],8c 1 and trityl-substituted ethylenediamine, 2 ligand in a one-pot reaction (Scheme 1) and characterized by 1H, 13C NMR and HRMS data (Fig. S1, S2 and S3, ESI†). The 1H NMR spectrum of 3 showed two aliphatic 1H signals at δ = 4.51 ppm (as triplet) and δ = 3.16 ppm (as broad singlet), which appeared at 2.83 ppm (as triplet) and 2.23 ppm (as triplet), respectively, in the 1H NMR spectrum of 2. The 13C NMR spectrum of 3 displayed three upfield resonance signals at 70.98, 48.68 and 44.47 ppm, which were ascribed to the sp3 carbon of the trityl group and two for the ethylenediamine carbons, respectively. From HRMS data, the major fragmented peak appeared as [M − Cl]+ at m/z 651.2009 and 2[M + H] at m/z 1376.3616, attributed to the dimeric species (Fig. S3, ESI†).
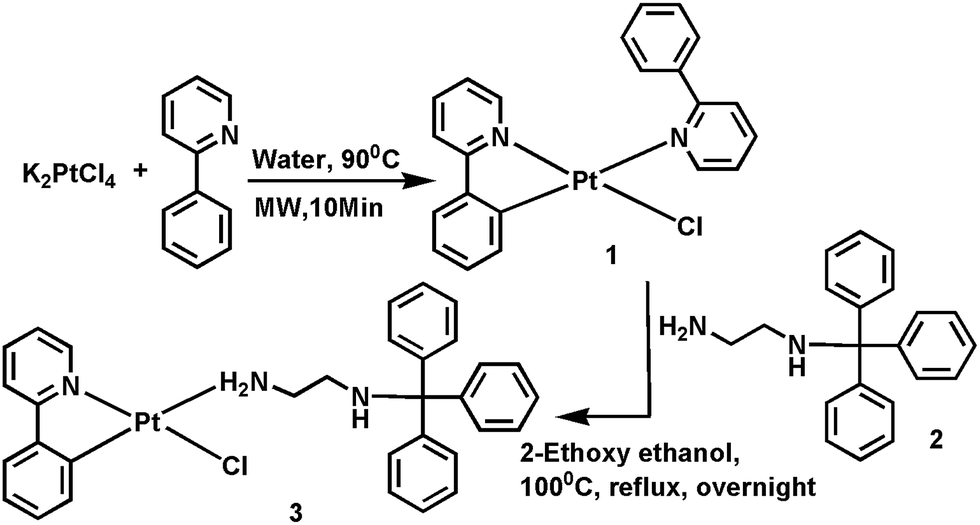 | ||
| Scheme 1 Synthetic routes of compounds 1, 2 and 3. | ||
The UV-Vis electronic absorption spectrum of 3 was recorded in dichloromethane, which exhibited three distinct absorption bands, that is, 300–350 nm, 352–420 nm and 425–465 nm (Fig. S4a, ESI†). Based on the nature of the spectrum and its similarity to earlier reports,3,4 the spectral bands were tentatively assigned to 1π–π*, 1MLCT and 3MLCT transitions, respectively (Fig. S4a, ESI†). These transitions were further supported by time-dependent density functional theory (TD-DFT), where the transition energy to the low-lying singlet states (S0–S1 and S0–S2) were calculated at 384 nm (oscillator strength, f = 0.0150) and 353 nm (f = 0.0056), respectively, and the lowest triplet states at 462 (S0–T1) and 390 nm (S0–T2), respectively (Table S1, ESI†). These two transitions, S0–S1 and S0–S2 were mainly comprised of HOMO → LUMO (94.0%) and (HOMO−2) → LUMO (97.0%) transitions, respectively (Table S1, ESI†). The electron distribution in S0–S1 and S0–S2 transitions, which is shown in Fig. S5b,† implies that there is a significant MLCT character (for LUMO, electron density found on Pt(II) for isovalue plot of 0.03 electrons/bohr3 was almost nil in comparison to the corresponding HOMO and HOMO−2) (Table S1, ESI†). The calculated % MLCTs were ∼56, ∼74 and ∼32 for S0–S1, S0–S2 and S0–T1 transitions, respectively. But the vibronic features observed at the experimentally obtained solution emission spectrum of 3 clearly indicated the presence of characteristic π–π* in the low-lying excited state (Fig. 3b). Hence, the low-lying excited state should comprise a mixed characteristic π–π* with MLCT. The solid-state UV-Vis absorption spectrum for crystalline sample 3 displayed a long-range relatively broader and stronger absorption band ranging from 300 nm to 450 nm as compared to the solution-phase absorption band. Following this stronger band, a low energy band, 470–510 nm, which was again followed by a long tail that extended up to 600 nm (Fig. S4a, ESI†). The low energy band at λmax = 487 nm was observed in solid state only, and predictably, not found in the solution absorbance spectrum. The nature of this band is ascribed to metal–metal-to-ligand charge transfer (3MMLCT) transitions (Fig. S4a, ESI†). Basically, the 3MMLCT state was generated from Pt(II) excimer.3,4 In the emission spectrum study, the low-energy vibronic structure (1313 cm−1) with λmax at 486 and 519 nm (in DCM) and a broad relatively low-energy emission spectrum with λmax at 570 nm (in solid state) were attributed to 3π–π* transitions mixed with 3MLCT and 3MMLCT transitions, respectively (Fig. S4c, ESI†). The time-resolved photoluminescence measurements (Fig. S6a–e, ESI†) were carried out of complex 3; lifetime was found to decrease linearly with increasing concentration (at c = 1 × 10−4 → 5 × 10−4 M, t = 116 ns → 94 ns), which was indicative of the presence of Pt(II)–Pt(II) interactions.
The pictorial representation of FMOs for the 3 complex is given in ESI (Fig. S5b, ESI†). The FMO analysis exhibited a dπ–pπ type of mixing in the lowest-lying HOMO, which was mainly confined on the Pt(II) and phenyl part of phenylpyridine (Fig. S5b and Table S2, ESI†). However, the LUMO mainly consisted of the π* orbital of the ligand phenylpyridine. Here, it is worth mentioning that the triphenyl ligand and the spacer part (ethylene diamine) do not provide any contribution to the HOMO–LUMO orbital.
A noticeable change in emission behaviour was observed when the 3 complex was ground in a mortar–pestle. In this case, the yellow emissive complex, 3 (λmax = 570 nm), was turned into the orange emissive complex, 3a (λmax = 600 nm). It reverted into the original 3 complex after recrystallizing the ground compound from DCM (Fig. 1). The change in such emission behaviour was attributed to mechanofluorochromism (MFC). Investigation of the MFC behaviour mechanism for 3 was carried out via powder X-ray diffraction (PXRD) and differential scanning calorimetry (DSC) experiments (Fig. S7 and S8†). The recorded PXRD spectrum showed sharp and intense peaks in the case of 3, which revealed the complex's crystalline packing mode.12 However, the PXRD spectrum of the ground sample exhibited low-intensity peaks that appeared as broad, indicating its amorphous nature. The PXRD data revealed that the change in the emission property was attributable to the change transition from crystalline to amorphous phase (Fig. S7, ESI†). This fact was further supported by the DSC experiment that showed a clear endothermic melting peak in both cases (199.9 and 170.0 °C), while a glass transition peak (t = 61 °C) followed by a broad exothermic re-crystallisation peak at ∼100 °C was observed in the ground sample case (Fig. S8, ESI†). A sharp difference in absorbance spectra of the orange-emitting complex, 3a, with the original yellow complex, 3, was observed (Fig. S4a, ESI†). Results for the complex 3a showed a prominent absorbance peak at ∼410 nm, along with a lowering absorbance as compared to the corresponding peak of 3 in solid-phase absorbance (absorbance ∼1.70 vs. ∼0.25). With the following prominent absorbance peak in 3a, a level-off tail was extended beyond 650 nm, but the absorbance of 3 was drastically reduced at longer wavelengths [425 (abs: 1.5)–525 nm (abs: 0.1)], after which the absorbance value tended to nearly zero (Fig. S4a, ESI†). In contrast to 3, there was no strong absorbance peak observed in the 470–510 nm range, a long and more red-shifted tail was observed and it was extended beyond 650 nm. The observed red-shifted absorbance and emission of 3a (in comparison to 3) would suggest MFC behaviour resulted in a stronger Pt–Pt stacking interaction.13 Hence, it was speculated that the two Pt(II) in 3 were moved in on grinding. The complex 3 was regenerated after recrystallizing of 3a from DCM.
 | ||
| Fig. 1 (a) Luminescent images of pristine complex 3 (yellow) after grinding one, 3a (orange), and after mixing of 3 with mesoporous silica (green) (photograph taken under 365 nm UV illumination). (b) Emission spectra as synthesized solid, 3 (black), ground solid, 3a (red = first time after grinding, blue = second time after grinding). (c) Maximum emission wavelength change upon repeated grinding–recrystallization cycles. | ||
To investigate the AIE property, the emission spectrum for 3 was recorded in different MeOH–H2O fractions (0–90%). The spectrum for 3 in MeOH exhibited a green emission with λmax at 485 nm and 518 nm, which did not show any significant enhancement in photoluminescence (PL) intensity while water fractions (fw), in the 10–60% range gradually increased. A sudden 39.4 times enhancement of PL intensity was observed at fw = 70% with red-shifted emission (λmax = 600 nm). The maximum PL intensity was achieved at fw = 90%, which was observed 62.1 times higher than its methanolic solution (Fig. 2). Aggregation formation was confirmed by the leveling-off of the absorbance spectrum (Fig. S10, ESI†) at higher water fractions. However, the sizes obtained from FESEM confirmed formation of aggregates at fw = 70% and fw = 90% with the size range of 114–150 nm and 85–104 nm, respectively (Fig. S11, ESI†). The suspended particles from fw = 90% was centrifuged, filtered and dried (3d). The absorbance and emission spectra of 3d were found to be totally different from the pristine 3 [λmax (emission) for 3, 570 nm and 3d, 600 nm] (Fig. S4, ESI†). But it was very similar to the emission profile observed for the ground form, 3a (Fig. S4d, ESI†). The XRD powder pattern of 3b supported its amorphous packing mode that was also similar to 3a (Fig. S7, ESI†). The different absorbance profile seen in 3d compared to 3a (Fig. S4b, ESI†) indicated differences in their ground-state geometries.
 | ||
| Fig. 2 (a) Emission spectra of 3 in a MeOH/water mixture (0–90%). (b) Plot of maximum emission intensity (I) and wavelength (λmax) of 3 versus water fraction; concentration of 3: 1 × 10−5 M. (c) Photographs of 3 in MeOH/water mixtures taken under UV illumination. (d) Image of solid thin film of 1 under UV light at λext 365 nm. | ||
The PL spectra for 3 were found to be nonemissive in common organic solvents because of active rotation of the propeller-shaped trityl group in the solution state. The PL spectrum of 3 was recorded in diverse polyethylene glycol (PEG)–MeOH mixtures (Fig. S9, ESI†). PL intensity was found to increase with increasing PEG fraction (fPEG). Maximum PL intensity was achieved at fPEG = 90%, which was 12 times higher than its pure methanolic solution. The PL enhancement with increasing viscous PEG revealed that the RIR may have blocked the nonradiative channels and opened up the radiative channels. The absolute quantum yield of 3 was measured to 12.35% and 0.4% in solid and solution states (in DCM), respectively, which supported the strong emissive property in the solid state.
Interestingly, the yellow-light emissive complex 3 (or orange emitting, 3a) was transformed into a green-emitting complex after mildly crushing it with mesostructured silica (MS silica) (Fig. 1a). Several experiments were carried out to investigate the cause underlying such an unusual property. The PL and absorption spectra of 3 (Fig. 3a, S12, ESI†) at a fixed concentration (10−4 M in DCM) were recorded in the presence of sequentially added different amounts of MS silica (0, 20, 40, 60, 80, 100 and 120 mg). In this case, the PL intensity and absorbance were gradually decreased with an increasing amount of MS silica. This experiment was indicative of encapsulation of 3 into the MS silica pores. The green-emitting MS silica (3b) was washed in a Soxhlet extraction apparatus with methanol and dichloromethane, respectively, for 72 h (24 h for MeOH and 48 h for DCM), which guaranteed removal of any adherents present on the MS silica surface. The washed MS silica displayed a bright green emission and the fact supporting impregnation of 3 into the void pores of MS silica. Differences in powder XRD patterns and FTIR spectra (Fig. 3c, S13, ESI†) of the bare MS silica with the 3b were not observed, which further supported the premise that the platinum(II) complex was encapsulated into the MS silica pores. The surface area of the encapsulated MS silica was measured by the BET experiment and the comparative surface areas of bare MS silica and encapsulated 3 were found at 750 and 387 m2 g−1, respectively (Table S3, ESI†), which confirmed the occupation of Pt(II) complex in the MS silica pores. The recorded HRTEM image of 3b showed the faded lattice fringes in comparison to the bare MS silica (Fig. S14, ESI†), which was further indicative of filling up void pores. 3b emitted green light that was similar to the green emissive 3 in DCM.
 | ||
| Fig. 3 (a) PL spectra of 3 with different amounts of MS silica in DCM (0–120 mg) with excitation at 365 nm. (b) Emission spectra of 3 in DCM (black) and 3b (red). (c) Powder XRD of bare MS silica (black), 3b (blue), and as synthesized complex 3 (green). | ||
Such observations support the premise that the framework of the MS silica remains unchanged under crushed condition. The difference in 10 nm blue-shifted (Fig. 3b) emission was observed for 3b (477 nm vs. 487 nm for 3), which could be explained by the exertion of pore size constraints over the encapsulants.14 Thus, the similar emissive nature of 3b with the emission profile in solution phase of 3 led us to conclude that the excimeric Pt(II) complex was dissociated into monomeric units under the crushed condition and thereby facilitated these smaller sized monomeric units to occupy positions in the empty pores.
In conclusion, a strategically designed AIE active platinum(II) complex was synthesized via a simple route. MFC behaviour of 3 demonstrated a transition from crystalline to amorphous state in aggregated form. In the molecular state, a strong Pt–Pt interaction was observed after crushing. A simple, dry approach to impregnate 3 into the mesostructured silica pores with concomitant sharp changes in emission color was observed. Investigations into the cause of such color change showed the dissociation of Pt(II)-excimer into monomeric units. The MS silica, encapsulated AIE active Pt(II) complex provides a space for tailor-made surface functionalization for important bio-applications.
Acknowledgements
We are grateful to the ‘DST, Govt. of India’ project SB/S1/IC-13/2014 and ‘CSIR, Govt. of India’ project no. 01/2551/12/EMR-II for financial support. R. K. R. acknowledges financial support of this research from DST, Govt. of India (Project Ref. No. SB/S1/PC-067/2013). We also acknowledge departmental ‘UGC-sponsored Special Assistance Programme’ and ‘DST-FIST’ for instrumental support. We are thankful to A. Roy Choudhury, IISER, Mohali, for providing the mass spectrum facility.Notes and references
- (a) V. H. Houlding and V. M. Miskowski, Coord. Chem. Rev., 1991, 111, 145 CrossRef CAS; (b) V. W.-W. Yam, K. M.-C. Wong and N. Zhu, J. Am. Chem. Soc., 2002, 124, 6506 CrossRef CAS PubMed; (c) W.-Y. Wong, G.-J. Zhou, X.-M. Yu, H.-S. Kwok and B.-Z. Tang, Adv. Funct. Mater., 2006, 16, 838 CrossRef CAS; (d) S. S. Pasha, P. Das, N. P. Rath, D. Bandyopadhyay, N. R. Jana and I. R. Laskar, Inorg. Chem. Commun., 2016, 67, 107 CrossRef CAS.
- (a) W.-Y. Wong and C.-L. Ho, Coord. Chem. Rev., 2009, 253, 1709 CrossRef CAS; (b) C. K. M. Chan, C.-H. Tao, H.-L. Tam, N. Zhu, V. W.-W. Yam and K.-W. Cheah, Inorg. Chem., 2009, 48, 7 CrossRef PubMed; (c) T. T. Feng, F. Q. Bai, L. M. Xie, Y. Tang and H. X. Zhang, RSC Adv., 2016, 6, 11648 RSC; (d) S. Seokhwan, L. H. Gyu, L. Nopl, R. Minwoo, K. Cheehun, L. Jihoon, A. Hogeun and C. Minchu, J. Nanosci. Nanotechnol., 2016, 16, 2028 CrossRef; (e) Y. Dai, H. Xiao, J. Liu, Q. Yuan, P. Ma, D. Yang, C. Li, Z. Cheng, Z. Hou, P. Yang and J. Lin, J. Am. Chem. Soc., 2013, 135, 18920 CrossRef CAS PubMed; (f) P. Yang, S. Gaib and J. Lin, Chem. Soc. Rev., 2012, 41, 3679 RSC.
- (a) K. M.-C. Wong and V. W.-W. Yam, Acc. Chem. Res., 2011, 44, 424 CrossRef CAS PubMed; (b) P. Pinter, H. Mangold, I. Stengel, I. Münster and T. Strassner, Organometallics, 2016, 35, 673 CrossRef CAS; (c) J. Zhanga, G. Daic, F. Wud, D. Lie, D. Gao, H. Jina, S. Chenf, X. Zhuf, C. Huanga and D. Han, J. Photochem. Photobiol., A, 2016, 316, 12 CrossRef; (d) M. Bachmann, D. Suter, O. Blacque and K. Venkatesan, Inorg. Chem., 2016, 55, 4733 CrossRef CAS PubMed; (e) J. J. Stace, P. J. Ball, V. Shingade, S. Chatterjee, A. Shiveley, W. L. Fleeman, A. J. Staniszewski, J. A. Krause and W. B. Connick, Inorg. Chim. Acta, 2016, 447, 98 CrossRef CAS.
- K. H.-Y. Chan, H. S. Chow, K. M.-C. Wong, M. C.-L. Yeung and V. W.-W. Yam, Chem. Sci., 2010, 1, 477 RSC.
- (a) M. L. Muro, C. A. Daws and F. N. Castellano, Chem. Commun., 2008, 6134 RSC; (b) T. Abe, T. Itakura, N. Ikeda and K. Shinozaki, Dalton Trans., 2009, 711 RSC; (c) J. S. Field, C. D. Grimmer, O. Q. Munro and B. P. Waldron, Dalton Trans., 2010, 39, 1558 RSC; (d) X. Zhang, J. Wang, J. Ni, L. Zhang and Z. Chen, Inorg. Chem., 2012, 51, 5569 CrossRef CAS PubMed; (e) J. R. Kumpfer, S. D. Taylor, W. B. Connick and S. J. Rowan, J. Mater. Chem., 2012, 22, 14196 RSC; (f) M. Krikorian, S. Liu and T. M. Swager, J. Am. Chem. Soc., 2014, 136, 2952 CrossRef CAS PubMed.
- C. H. Huang, F. Y. Li and W. Huang, Introduction to Organic Light-Emitting Materials and Devices, Press of Fudan University, Shanghai, 2005 Search PubMed.
- Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361 RSC.
- (a) S. Liu, H. Sun, Y. Ma, S. Ye, X. Liu, X. Zhou, X. Mou, L. Wang, Q. Zhao and W. Huang, J. Mater. Chem., 2012, 22, 22167 RSC; (b) H. Honda, Y. Ogawa, J. Kuwabara and T. Kanbara, Eur. J. Inorg. Chem., 2014, 1865 CrossRef CAS; (c) S. S. Pasha, P. Alam, S. Dash, G. Kaur, D. Banerjee, R. Chowdhury, N. Rath, A. R. Choudhury and I. R. Laskar, RSC Adv., 2014, 4, 50549 RSC.
- Z. Li, J. C. Barnes, A. Bosoy, J. F. Stoddart and J. I. Zink, Chem. Soc. Rev., 2012, 41, 2590 RSC.
- (a) O. H. Park, S. Y. Seo, J. I. Jung, J. Y. Bae and B. S. Bae, J. Mater. Res., 2003, 18, 5 Search PubMed; (b) Q. G. Meng, P. Boutinaud, A. C. Franville, H. J. Zhang and R. Mahiou, Microporous Mesoporous Mater., 2003, 65, 127 CrossRef CAS; (c) H. M. Yu, H. D. Liang and Z.-Z. Yan, J. Coord. Chem., 2011, 64, 440 CrossRef CAS.
- (a) P. Alam, M. Karanam, A. Roy Choudhury and I. R. Laskar, Dalton Trans., 2012, 41, 9276 RSC; (b) P. Alam, P. Das, C. Climentc, M. Karanam, D. Casanovac, A. R. Choudhury, P. Alemany, N. R. Jana and I. R. Laskar, J. Mater. Chem. C, 2014, 2, 5615 RSC.
- Y. Q. Dong, J. W. Y. Lam and B. Z. Tang, J. Phys. Chem. Lett., 2015, 6, 3429 CrossRef CAS PubMed.
- (a) T. Abe, T. Itakura, N. Ikeda and K. Shinozaki, Dalton Trans., 2009, 711 RSC; (b) J. Ni, X. Zhang, Y. Wu, L. Zhang and Z. Chen, Chem.–Eur. J., 2011, 17, 1171 CrossRef CAS PubMed.
- H. Zhang, Y. Sun, K. Ye, P. Zhang and Y. Wang, J. Mater. Chem., 2005, 15, 3181 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra17273c |
| This journal is © The Royal Society of Chemistry 2016 |