
Fe3O4@SiO2/Schiff base/Pd complex as an efficient heterogeneous and recyclable nanocatalyst for chemoselective N-arylation of O-alkyl primary carbamates†
A. R. Sardarian*,
M. Zangiabadi and
I. Dindarloo Inaloo
Department of Chemistry, College of Sciences, Shiraz University, Shiraz 71946 84795, Iran. E-mail: sardarian@shirazu.ac.ir
First published on 13th September 2016
Abstract
An efficient, heterogeneous and cost effective method has been developed using an Fe3O4@SiO2/Schiff base/Pd complex as a magnetic and easily recyclable nanocatalyst for rapid and effective N-arylation of carbamates in good to excellent yield. The catalyst can be easily recovered and reused over six runs without significant decrease in the activity. Further highlights of this protocol are operational simplicity, versatility and relatively short reaction times.
Introduction
Due to the biological activity of compounds which have a nitrogen atom in their structures and the importance of finding new ways to effectively synthesise such molecules, C–N coupling reactions have been a topic of interest in the last two decades.1–5 Although in most of these methods almost all nitrogen sources such as amines,6 amides,7 indoles,8 azoles9 and amino acids10 have been used for the creation of C–N bonds, only a few examples of the application of carbamates11–13 as another nitrogen source have been reported in the literature.Carbamates are an important class of compounds in organic chemistry because of their considerable biological activities,14–16 their use in the synthesis of polymers17 and as protecting groups in the synthesis of organic compounds.18
Carbamate skeletons are frequently found in a variety of biologically active molecules, especially in medicinal chemistry and pharmaceuticals. The most important group of carbamate derivatives are unsymmetrical compounds (Fig. 1), known to be widely used in modern drug design.
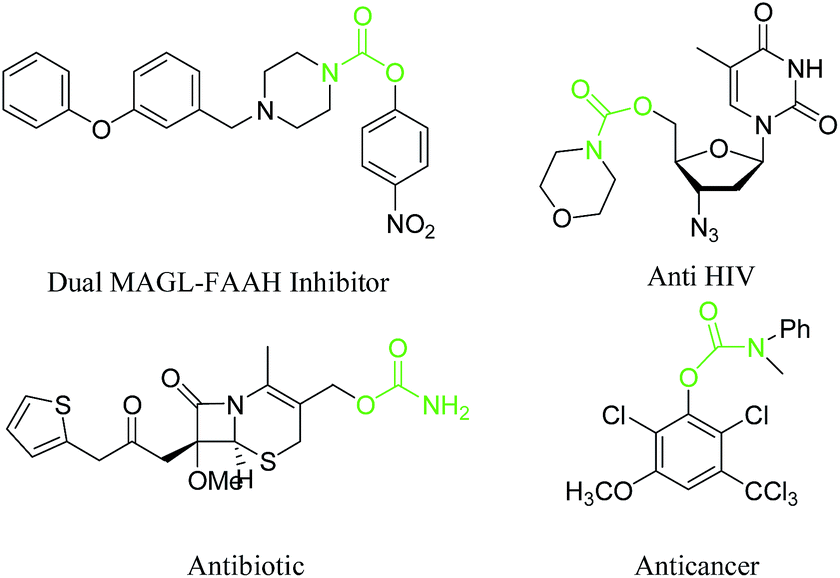 | ||
| Fig. 1 The chemical structure of some important and biological active carbamates. | ||
As the result, there are several reports for synthesis of secondary and tertiary carbamates including the commercial procedure which uses the phosgenation of amines,19 the reaction of dimethylcarbonates with amines,20 the reductive carbonylation of nitro compounds21,22 and by the reaction of CO2 with amines and alcohols.23,24
Aryl carbamates can be produced by the reductive carbonylation of nitroarenes.25 They are also prepared by the reaction of isocyanate with aryl halides26,27 and aryl boronic acids28 in the presence of transition metal complexes of Pd and Cu. In one report they have been produced by using C–H functionalization method.29
Magnetic nanocatalysts have gained great attention in the recent years. They can be easily removed from the reaction mixture by an external magnetic field and reused several times. This operational simplicity made them very useful and efficient catalysts in a wide range of organic reactions.30–33
To the best of our knowledge there are only few reports which carry out the C–N coupling reaction of carbamates directly to afford aryl carbamates.12,13,34 These protocols suffer from some major drawbacks, such as limitation in the scope of reaction,13,28 using nonrecyclable homogeneous catalyst,13,28 long reaction times and using expensive ligands for making the catalyst.13,27
To overcome these shortcomings, herein we introduce a magnetic Pd nanocatalyst which is able to catalyze efficiently the C–N coupling reaction among primary O-alkyl carbamates and aryl halides and can be easily removed from the reaction by an external magnetic field and reused.
Results and discussion
The magnetic nanocatalyst was synthesized through the protocol which was reported in 2014 by Esmaeilpour et al.35 Scheme 1 concisely describes the synthetic procedure of the catalyst. | ||
| Scheme 1 Synthetic procedure of Fe3O4@SiO2/Pd(II) complex. | ||
IR spectrum, XRD pattern and DLS image of the catalyst are shown in Fig. 3. According to IR spectrum, peaks at 1100, 1618, 2973 and 3414 are related to Si–O–Si, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N, C–H and O–H groups respectively. In addition, the characteristic peaks of Fe3O4 are present in the XRD pattern of catalyst at 2θ = 30.1°, 35.4°, 43.1°, 53.4°, 57° and 62.6°, which were marked respectively by their indices (220), (311), (400), (422), (511) and (440). Moreover, characteristic peaks of Pd(0) cannot be observed in XRD pattern, which indicates that the Pd particles on the core–shell catalyst are Pd(II). DLS image of the catalyst shows the distribution of particles regarding size in which the most frequent size of particles was 32 nm. Also, the ICP analysis demonstrated that the Pd loading on the catalyst was 0.26 mmol g−1. These results are consistent with the reported article and confirm the structure of the catalyst.
N, C–H and O–H groups respectively. In addition, the characteristic peaks of Fe3O4 are present in the XRD pattern of catalyst at 2θ = 30.1°, 35.4°, 43.1°, 53.4°, 57° and 62.6°, which were marked respectively by their indices (220), (311), (400), (422), (511) and (440). Moreover, characteristic peaks of Pd(0) cannot be observed in XRD pattern, which indicates that the Pd particles on the core–shell catalyst are Pd(II). DLS image of the catalyst shows the distribution of particles regarding size in which the most frequent size of particles was 32 nm. Also, the ICP analysis demonstrated that the Pd loading on the catalyst was 0.26 mmol g−1. These results are consistent with the reported article and confirm the structure of the catalyst.
The magnetic property of the catalyst is demonstrated in Fig. 2. According to this figure, the catalyst has good dispersity in the reaction media and it can be easily separated from the mixture by an external magnetic field.
 | ||
| Fig. 2 (a) Good dispersity and (b) easy separation of the catalyst by an external magnetic field. | ||
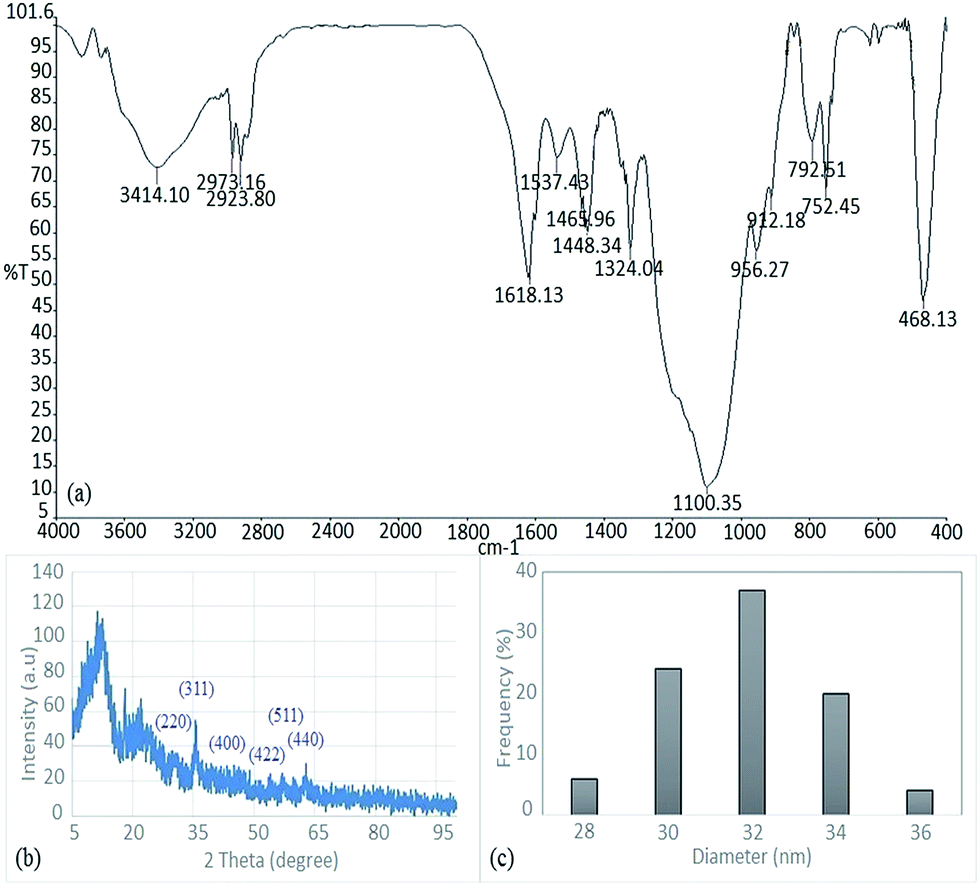 | ||
| Fig. 3 (a) FT-IR spectrum, (b) XRD pattern and (c) DLS image of the catalyst. | ||
Fig. 4 illustrates the FE-SEM image of the Pd nanocatalyst. The spherical morphology of the catalyst is clearly shown in this figure.
 | ||
| Fig. 4 FE-SEM image of the fresh catalyst. | ||
Among the last reports for arylation of carbamates, the use of homogenous catalysts seems to be the only practical method, which suffers from the above mentioned drawbacks. In this work, for the first time, we report an efficient magnetic heterogeneous Pd nanocatalyst for N-arylation of primary O-alkyl carbamates, which can be easily recovered by an external magnetic field and reused, with aryl halides.
Initially, propyl carbamate and iodobenzene were selected as the model substrates and then their reaction in the presence of Fe3O4@SiO2/Schiff base/Pd complex (0.14 wt%), as a magnetic nanocatalyst in the common solvents, such as DMF, DMSO, dioxane, toluene, dichloromethane, acetonitrile and ethanol, and bases, such as CsCO3, K2CO3, NaOBut and triethylamine at different temperatures were explored. The corresponding results were summarized in Table 1. According to these data, the reaction showed the best efficiency when the model reaction was run in toluene and in the presence of 1.5 mmol NaOBut and 0.055 wt% (0.02 g) the Pd nanocatalyst at 100 °C for 2.5 hours (Table 1, entry 14).
| Entry | Solvent | Base | Catalyst (wt%) | Temp. (°C) | Yield (%) |
|---|---|---|---|---|---|
| a Reaction conditions: propyl carbamate (1 mmol), iodobenzene (1 mmol), base (1.5 mmol) and catalyst in 2 mL solvent for 12 h. | |||||
| 1 | Toluene | NaOBut | 0.14 | 100 | 92 |
| 2 | DMF | NaOBut | 0.14 | 100 | Trace |
| 3 | CH3CN | NaOBut | 0.14 | Reflux | 0 |
| 4 | CH2Cl2 | NaOBut | 0.14 | Reflux | 0 |
| 5 | Ethanol | NaOBut | 0.14 | Reflux | 0 |
| 6 | Dioxane | NaOBut | 0.14 | 100 | 78 |
| 7 | DMSO | NaOBut | 0.14 | 100 | 20 |
| 8 | Toluene | K2CO3 | 0.14 | 100 | 10 |
| 9 | Toluene | CsCO3 | 0.14 | 100 | 53 |
| 10 | Toluene | Et3N | 0.14 | 100 | 0 |
| 11 | Toluene | NaOBut | 0.14 | 25 | Trace |
| 12 | Toluene | NaOBut | 0.14 | 50 | 38 |
| 13 | Toluene | NaOBut | 0.14 | 120 | 88 |
| 14 | Toluene | NaOBut | 0.055 | 100 | 94 |
| 15 | Toluene | NaOBut | 0.19 | 100 | 87 |
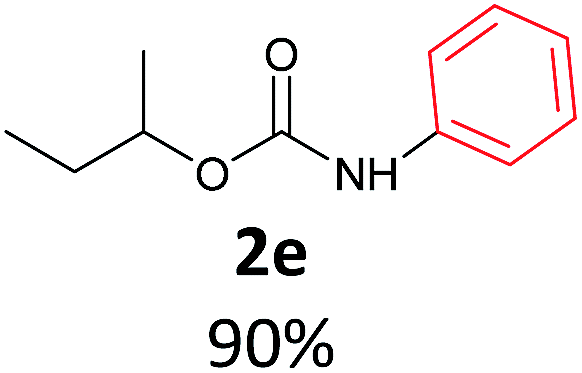
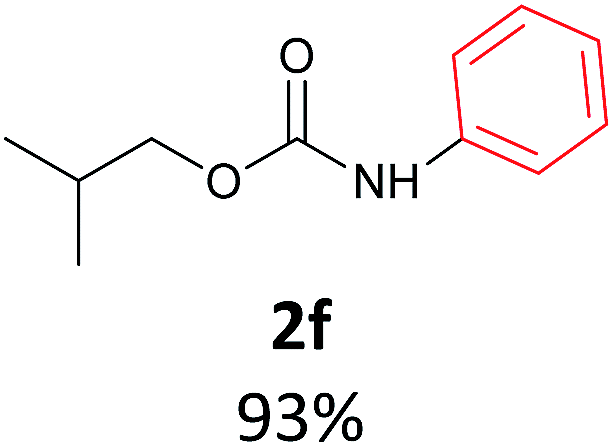
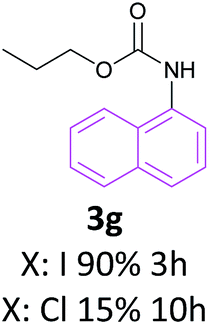
With the optimized reaction parameters in hand, initially a variety of O-alkyl primary carbamates were treated with aryl iodide in toluene and in the presence of NaOBut and the catalyst at 100 °C to afford the corresponding N-aryl carbamates in good to excellent yields. The results were summarized in Table 2. A variety of carbamates were coupled with iodobenzene to afford the corresponding N-aryl carbamates in good to excellent yields. As it is illustrated in Table 2, in addition of the simple O-alkyl primary carbamates, this procedure was effective also for tert-butyl carbamate (Table 2, entry 2g) and benzyl carbamate (Table 2, entry 2i) which are widely used as the protecting groups in organic synthesis. Also, sterically hindered carbamates carried out the reaction very well (Table 2, entry 2j). In the case of O-aryl carbamates, although they underwent the reaction, as it is reported by Buchwald et al. they are unstable and cannot isolated and purified by chromatographic procedures.27
|
||
|---|---|---|
| a Reaction conditions: carbamate (1 mmol), iodobenzene (1 mmol), sodium tert-butoxide (1.5 mmol), catalyst (0.055 wt%) in 2 mL toluene at 100 °C for 2.5 h. | ||
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
||

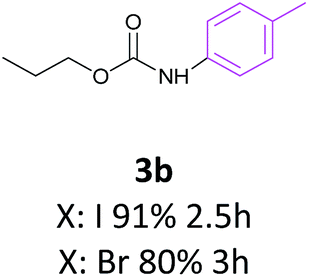
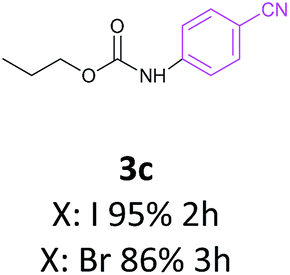
Also the coupling reaction of O-propyl carbamate with the other aryl iodides was explored and the results were represented in Table 3. According to these data, aryl iodides with electron donating and electron withdrawing substituents carried out the C–N coupling reaction efficiently and gave the corresponding secondary carbamates in excellent yields, however, aryl halides bearing electron withdrawing groups gave slightly higher yields (Table 3, entries 3c, 3d and 3e). Moreover, 3-iodothiophen, as a heterocyclic aromatic iodide, reacted with O-propyl carbamate in the presence of catalyst under optimized condition and produced the desired product in high yield (Table 3, entry 3f).
|
||
|---|---|---|
| a Reaction conditions: O-Propyl carbamate (1 mmol), aryl halide (1 mmol), sodium tert-butoxide (1.5 mmol), catalyst (0.055 wt%) in 2 mL toluene at 100 °C. | ||
 |
 |
 |
 |
 |
 |
 |
 |
|

These interesting results encouraged us to study the activity of aryl bromides and aryl chlorides under the optimized condition of C–N coupling (toluene as solvent NaOBut as base in 100 °C) in the presence of Fe3O4@SiO2/Schiff base/Pd complex. The data obtained from this study were also exhibited in Table 3. These results showed that aryl bromides, as the same as aryl iodides, carried out the C–N coupling reaction with O-propyl carbamate efficiently however with slightly lower yields than aryl iodides. The reaction between O-propyl carbamate and inactive aryl chlorides were occurred in longer reaction times and lower yields compared to aryl iodides and aryl bromides and the starting carbamate mainly remained (Table 3, entries 3a and 3g). While the yields are low for aryl chlorides in this protocol, these substrates are completely inactive in the majority of the methods reported in the literature.
To check chemoselectivity of the protocol, the reaction between 1-bromo-4-iodobenzene with O-propyl carbamate was investigated under optimized condition. The result of this study indicated that the C–N coupling reaction has been taken place only on the carbon atom bearing iodine and only propyl 4-bromophenylcarbamate was prepared (Table 4, entry 1). Then the same reaction was performed between 1-chloro-4-iodobenzene and O-propyl carbamate. In this case only propyl 4-chlorophenylcarbamate was obtained (Table 4, entry 2). Finally, the reaction of O-propyl carbamate with 1-bromo-4-chlorobenzene was examined and as it is mentioned in Table 4 only the propyl 4-chlorophenylcarbamate was observed in 88% yield (Table 4, entry 3). These results indicate excellent chemoselectivity and the C–N coupling reaction is occurred on the carbon atom bears less electronegative halogen atom.
|
||||
|---|---|---|---|---|
| Entry | Substrate | Product 1 (yield%) | Product 2 (yield%) | Product 3 (yield%) |
| 1 |  |
91 | — | 0 |
| 2 |  |
— | 93 | 0 |
| 3 |  |
0 | 88 | — |

In order to compare the reactivity among carbamates and amides, the C–N coupling reaction between O-propyl primary carbamate and propanamide with iodobenzene was examined. For this purpose, 1 mmol of iodobenzene was reacted with 1 mmol of O-propyl carbamate and 1 mmol of propanamide in the presence of catalyst under the optimized conditions. As it is depicted in Scheme 2, analysis of the reaction mixture appeared that propanamide remained unchanged and only O-propyl carbamate underwent the C–N coupling selectively. This could be due to the stronger nucleophilicity of nitrogen atom in carbamates in comparison to amides, which is important in Pd catalyzed C–N coupling reactions.36
 | ||
| Scheme 2 Comparing the reactivity between O-propyl carbamate and propanamide toward C–N coupling reaction. | ||
The recyclability of the catalyst was explored on the reaction between iodobenzene and O-propyl carbamates. After completion of the reaction, the catalyst was removed from the mixture by an external magnetic field and reused in the next same reaction. The data presented in Fig. 5 show that the Pd nanocatalyst is reusable after six runs without significant losing in activity.
 | ||
| Fig. 5 Recyclability of the catalyst. | ||
Fig. 6 represents the XRD pattern and FE-SEM image of the recycled catalyst after sixth run. This figure shows that characteristic peaks of Fe3O4 are present well and also there is no observation of Pd(0) species in the XRD pattern of the catalyst after recycling. Also the FE-SEM image demonstrates that Pd nanocatalyst keeps its spherical shape but with little agglomeration, which is believed to be the main cause of yield decreasing.
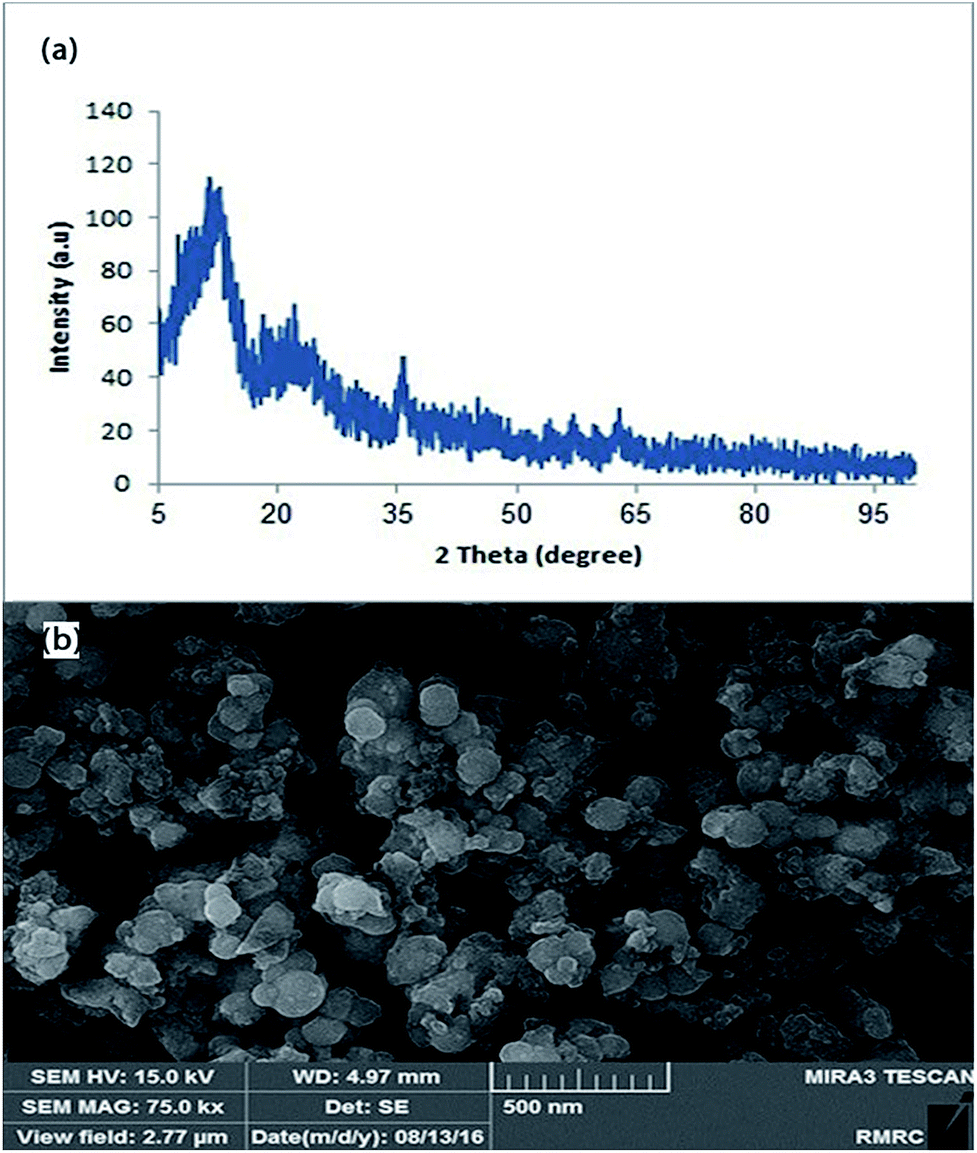 | ||
| Fig. 6 (a) XRD pattern and (b) FE-SEM image of the catalyst after sixth run. | ||
To find responsible Pd species in the C–N coupling reaction, hot filtration test was performed on the reaction of iodobenzene and O-propyl carbamates after six runs. When the reaction was half done the Pd nanocatalyst were removed from the reaction mixture by an external magnetic field. Then the reaction mixture was allowed to proceed at the reaction temperature. Only a very small conversion occurred after the removal of catalyst from the reaction moiety (about 2%). These results showed that only a few species of Pd exist in the solution phase and the main responsibility of the reaction progress is due to the heterogeneous catalyst. Inductively coupled plasma (ICP) analysis was also performed for the fresh catalyst and the catalyst after one and after six runs to determine the amount of palladium leaching. According to this test the amount of palladium on the fresh catalyst was obtained to be 0.26 mmol g−1. ICP analysis after one run showed only 0.3% for palladium leaching and after six runs it was determined to be 3.6%. These data along with those for hot filtration test, confirm the high heterogeneity and stability of the catalyst.
Experimental
General
All chemicals were purchased from the Merck, Flucka and Aldrich Chemical Companies in high purity. The products were characterized by comparison of their spectral and physical data such as NMR, FT-IR, MS, CHNS and melting point with available literature data. 1H and 13C-NMR spectra were recorded with Bruker Avance DPX 250 MHz instruments with Me4Si or solvent resonance as the internal standard. 1H NMR spectroscopic data are reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, quint = quintet, sext = sextet, sept = septet, br = broad, m = multiplet), coupling constants (Hz), and integration. Fourier transform infrared (FTIR) spectra were obtained using a Shimadzu FT-IR 8300 spectrophotometer. The hydrodynamic size of the particles was measured by dynamic light scattering (DLS) techniques, using a HORIBA-LB550 particle size analyzer. Pd loading and leaching test was carried out with an inductively coupled plasma (ICP) analyzer (Varian, vista-pro). Determination of the purity of the substrate and monitoring of the reaction were accomplished by thin-layer chromatography (TLC) on a silica-gel polygram SILG/UV 254 plates.To a mixture of alcohol (1.0 mmol) and potassium cyanate (1.5 mmol) in a mortar, DBSA (1.5 mmol) was added and the mixture was pulverized until a uniform mixture was formed. Then the mixture was kept in an oven at 60 °C for 1 hour. After completion of the reaction, CH2Cl2 (15 mL) and saturated aqueous solution of NaHCO3 (15 mL) were added to the resulting powder and organic layer was extracted, washed with water (3 × 15 mL), dried over anhydrous Na2SO4 and concentrated to afford the final product. Finally, by recrystallization from diethyl ether pure product was obtained.
1H-NMR (250 MHz, 298 K, CDCl3) δ ppm; 0.91 (t, J = 7.5 Hz, 3H), 1.63 (sext, J = 7.5 Hz, 2H), 2.50 (s, 3H), 4.08 (t, J = 7.5 Hz, 2H), 6.95 (s,br, 1H, NH), 7.43 (d, J = 7.5 Hz, 2H), 7.85 (d, J = 7.5 Hz, 2H). 13C-NMR (63 MHz, 298 K, CDCl3), δ ppm; 10.31, 22.18, 26.38, 67.22, 117.54, 129.87, 130.88, 142.51, 153.21, 200.49.
Conclusion
In conclusion, we report here a new heterogeneous method for the arylation of various carbamates with aryl halides. The procedure is interesting due to some advantageous such as high yields, using a heterogeneous catalyst, easy separation and recyclability of catalyst and relatively short reaction times. The catalyst can be easily removed from the reaction mixture by an external magnetic field and reused for at least 6 runs.Acknowledgements
Authors gratefully acknowledge the financial support of this work by the research council of Shiraz University.References
- D. S. Surry and S. L. Buchwald, Chem. Sci., 2011, 2, 27–50 RSC.
- A. R. Muci and S. L. Buchwald, in Cross-Coupling Reactions, Springer, 2002, pp. 131–209 Search PubMed.
- J. F. Hartwig, Angew. Chem., Int. Ed., 1998, 37, 2046–2067 CrossRef CAS.
- M. S. Driver and J. F. Hartwig, J. Am. Chem. Soc., 1997, 119, 8232–8245 CrossRef CAS.
- J. Bariwal and E. Van der Eycken, Chem. Soc. Rev., 2013, 42, 9283–9303 RSC.
- J. Jiao, X.-R. Zhang, N.-H. Chang, J. Wang, J.-F. Wei, X.-Y. Shi and Z.-G. Chen, J. Org. Chem., 2011, 76, 1180–1183 CrossRef CAS PubMed.
- J. D. Hicks, A. M. Hyde, A. M. Cuezva and S. L. Buchwald, J. Am. Chem. Soc., 2009, 131, 16720–16734 CrossRef CAS PubMed.
- D. T. Ziegler, J. Choi, J. M. Muñoz-Molina, A. C. Bissember, J. C. Peters and G. C. Fu, J. Am. Chem. Soc., 2013, 135, 13107–13112 CrossRef CAS PubMed.
- L. Zhu, P. Guo, G. Li, J. Lan, R. Xie and J. You, J. Org. Chem., 2007, 72, 8535–8538 CrossRef CAS PubMed.
- K. K. Sharma, S. Sharma, A. Kudwal and R. Jain, Org. Biomol. Chem., 2015, 13, 4637–4641 CAS.
- A. A. Trabanco, J. A. Vega and M. A. Fernández, J. Org. Chem., 2007, 72, 8146–8148 CrossRef CAS PubMed.
- S. Bhagwanth, A. G. Waterson, G. M. Adjabeng and K. R. Hornberger, J. Org. Chem., 2009, 74, 4634–4637 CrossRef CAS PubMed.
- L. Qin, H. Cui, D. Zou, J. Li, Y. Wu, Z. Zhu and Y. Wu, Tetrahedron Lett., 2010, 51, 4445–4448 CrossRef CAS.
- T. T. Wu, J. Huang, N. D. Arrington and G. M. Dill, J. Agric. Food Chem., 1987, 35, 817–823 CrossRef CAS.
- R. K. Pandey, S. P. Dagade, M. K. Dongare and P. Kumar, Synth. Commun., 2003, 33, 4019–4027 CrossRef CAS.
- W. Li, J. Li, Y. Wu, J. Wu, R. Hotchandani, K. Cunningham, I. McFadyen, J. Bard, P. Morgan, F. Schlerman, X. Xu, S. Tam, S. J. Goldman, C. Williams, J. Sypek and T. S. Mansour, J. Med. Chem., 2009, 52, 1799–1802 CrossRef CAS PubMed.
- B. S. Lee, B. C. Chun, Y.-C. Chung, K. I. Sul and J. W. Cho, Macromolecules, 2001, 34, 6431–6437 CrossRef CAS.
- X. Y. Xiao, K. Ngu, C. Chao and D. V. Patel, J. Org. Chem., 1997, 62, 6968–6973 CrossRef CAS.
- H. Eckert and B. Forster, Angew. Chem., Int. Ed. Engl., 1987, 26, 894–895 CrossRef.
- N. Lucas, A. P. Amrute, K. Palraj, G. V. Shanbhag, A. Vinu and S. B. Halligudi, J. Mol. Catal. A: Chem., 2008, 295, 29–33 CrossRef CAS.
- F. Paul, J. Fischer, P. Ochsenbein and J. A. Osborn, Organometallics, 1998, 17, 2199–2206 CrossRef CAS.
- F. Ragaini, M. Gasperini and S. Cenini, Adv. Synth. Catal., 2004, 346, 63–71 CrossRef CAS.
- M. Abla, J.-C. Choi and T. Sakakura, Chem. Commun., 2001, 2238–2239, 10.1039/b106201h.
- M. Honda, S. Sonehara, H. Yasuda, Y. Nakagawa and K. Tomishige, Green Chem., 2011, 13, 3406–3413 RSC.
- Q. Yang, A. Robertson and H. Alper, Org. Lett., 2008, 10, 5079–5082 CrossRef CAS PubMed.
- X. Yang, Y. Zhang and D. Ma, Adv. Synth. Catal., 2012, 354, 2443–2446 CrossRef CAS.
- E. V. Vinogradova, N. H. Park, B. P. Fors and S. L. Buchwald, Org. Lett., 2013, 15, 1394–1397 CrossRef CAS PubMed.
- E. Kianmehr and M. H. Baghersad, Adv. Synth. Catal., 2011, 353, 2599–2603 CrossRef CAS.
- P. K. Chikkade, Y. Kuninobu and M. Kanai, Chem. Sci., 2015, 6, 3195–3200 RSC.
- M. J. Jacinto, O. H. Santos, R. F. Jardim, R. Landers and L. M. Rossi, Appl. Catal., A, 2009, 360, 177–182 CrossRef CAS.
- M. J. Jin and D. H. Lee, Angew. Chem., Int. Ed., 2010, 49, 1119–1122 CrossRef CAS PubMed.
- T. Zeng, L. Yang, R. Hudson, G. Song, A. R. Moores and C.-J. Li, Org. Lett., 2010, 13, 442–445 CrossRef PubMed.
- M. Esmaeilpour, A. R. Sardarian and J. Javidi, Appl. Catal., A, 2012, 445, 359–367 CrossRef.
- J. F. Hartwig, M. Kawatsura, S. I. Hauck, K. H. Shaughnessy and L. M. Alcazar-Roman, J. Org. Chem., 1999, 64, 5575–5580 CrossRef CAS PubMed.
- M. Esmaeilpour, A. R. Sardarian and J. Javidi, J. Organomet. Chem., 2014, 749, 233–240 CrossRef CAS.
- I. P. Beletskaya and A. V. Cheprakov, Organometallics, 2012, 31, 7753–7808 CrossRef CAS.
- A. R. Sardarian and I. D. Inaloo, RSC Adv., 2015, 5, 76626–76641 RSC.
- N. Uhlig and C. J. Li, Chem.–Eur. J., 2014, 20, 12066–12070 CrossRef CAS PubMed.
- X. Zhang, H. Jing and G. Zhang, Synth. Commun., 2010, 40, 1614–1624 CrossRef CAS.
- E. P. Papadopoulos and C. M. Schupbach, J. Org. Chem., 1979, 44, 99–104 CrossRef CAS.
- M. Nardi, N. H. Cano, P. Costanzo, M. Oliverio, G. Sindona and A. Procopio, RSC Adv., 2015, 5, 18751–18760 RSC.
- C. Stock and R. Brückner, Adv. Synth. Catal., 2012, 354, 2309–2330 CrossRef CAS.
- S. Shulman and J. A. Griepentrog, Microchem. J., 1962, 6, 179–197 CrossRef CAS.
- J. Attaway, R. Wolford, G. Alberding and G. Edwards, Anal. Chem., 1962, 34, 671–673 CrossRef CAS.
- P. Sah and K. Y. Tao, Recl. Trav. Chim. Pays-Bas, 1939, 58, 12–16 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Copies of FT-IR, 1H and 13C-NMR and MS spectra for all compounds. See DOI: 10.1039/c6ra17268g |
| This journal is © The Royal Society of Chemistry 2016 |