
Immobilization of soybean peroxidase on silica-coated magnetic particles: a magnetically recoverable biocatalyst for pollutant removal
Maria C. Silva*a,
Juliana A. Torresb,
Francisco G. E. Nogueirac,
Tássia S. Tavaresb,
Angelita D. Corrêab,
Luiz C. A. Oliveiraa and
Teodorico C. Ramalho*b
aDepartment of Chemistry, Universidade Federal de Minas Gerais, 31270-901 Belo Horizonte, Brazil. E-mail: crisiria@yahoo.com.br
bDepartment of Chemistry, Universidade Federal de Lavras, Lavras, 37200-000, Brazil
cDepartment of Chemistry Engineering, Universidade Federal de São Carlos, 13565-905 São Carlos, Brazil
First published on 31st August 2016
Abstract
In this work we investigated the enzymatic degradation of ferulic acid, a model pollutant, by free and immobilized soybean peroxidase. With the aim of developing greener catalysts, we proposed the synthesis of a magnetic catalyst prepared via immobilization of soybean peroxidase onto a magnetic nanosupport by covalent attachment. The immobilization of soybean peroxidase was carried out using magnetite nanoparticles modified with amino groups as support. The magnetite particles were characterized before and after chemical modification by XRD, SEM and TEM analysis. The characterization data indicated that the Fe3O4–SiO2 nanoparticles were successfully synthetized. The high immobilization yield was obtained in only 1 hour of reaction (89.23%). The resulting nanobiocatalyst (enzyme load 5.25 U) was able to remove 99.67 ± 0.10% of ferulic acid in comparison to 57.67 ± 0.27% for free enzyme under the same reaction conditions. The immobilized peroxidase could easily be separated under a magnetic field and reused. On the basis of these results, we concluded that the prepared magnetic nanoparticles can be considered a high-performance nanocatalyst for environmental remediation.
Introduction
Concerning environmental issues, the development of highly efficient catalysts constitutes one important approach to minimize the effect of pollutants in wastewaters. In this context, the use of enzymes, which are versatile and specific catalysts, has constantly increased over time due to the peculiar properties of this class of proteins.1Peroxidases, like horseradish peroxidase (HRP) and soybean peroxidase (SP) are efficient catalysts for the removal of phenols from industrial wastewaters.2–6 These enzymes are able to catalyse the oxidation of phenols in the presence of H2O2, producing free radicals. The generated free radicals react with each other in a non-enzymatic process to form polymers, which can be easily separated from aqueous medium.3,4
Ferulic acid, a phenolic compound, is considered a high-priority environmental pollutant due to its high resistance to conventional biological oxidation. Ferulic acid is a lignin model compound and can be found in many industrial effluents and residues, including those produced in wine-distilleries, olive oil processing industries, pulp paper processing and others.7,8
The short catalytic lifetime of the enzyme is one significant drawback in enzymatic technology, mainly to phenolic compounds degradation, due to the inactivation of the enzyme induced by the polymerization process.9 The enzyme immobilization is an effective way to overcome this limitation. Besides improving stability and promoting significant enhancement in the overall process efficiency, the biocatalyst can be reused and recovered at the end of the process and be used in continuous processes.1
The immobilization strategies include encapsulation or entrapment, physical adsorption and covalent attachment.10,11 The most popular enzyme immobilization method is the physical adsorption. This method is characterized by its simplicity, because previous functionalization of the carrier surface is typically not required. Denaturation and/or deactivation of the enzyme can be avoided and, thus, retention of catalytic activity of the immobilized enzyme is very high.12,13 Moreover, the enzyme stability is significantly improved since this process makes use of the physical interactions generated between the carrier and enzyme that include van der Waals forces, ionic interactions and hydrogen bonding. The binding are rather weak and, what is important, typically are does not change the native structure of the enzyme. This prevents the active sites of the enzyme from disturbing and allows the enzyme to retain its activity. Physical bonding is generally too weak to keep the enzyme fixed to the carrier and is prone to leaching of the enzyme. Besides that, the physical adsorption is quite liable to changes under certain conditions such as pH, temperature and ionic strength of the buffer.14
Therefore, adsorption methods are simple and provide a high enzyme loading and a significant stability improvement, however, they are accompanied by risks derived from non-specific protein binding and enzyme loss during operation. An attractive solution for this problem is covalent attachment, which is the most effective approach to prevent detachment of the enzyme from support during operation.11,15
Regarding to supports commonly used for enzyme immobilization, the porous materials can afford high enzyme loading, however, nonporous nanoparticles, such magnetite, compared to these materials have no external diffusion problems, making them more competitive especially for large scale industrial usage in solid–liquid systems.16,17 Besides that, the enzyme bound nanoparticles show Brownian movement, when dispersed in aqueous solutions showing that the enzymatic activities are comparatively better than that of the unbound enzyme.18 In spite of associated advantages of immobilized enzyme onto magnetic nanoparticles, it has also some disadvantages including lowered activity, conformational change of the enzyme, possibility of enzyme denaturation, changes in properties, mass transfer limitations, and lowered efficacy against insoluble substrates.19
Magnetic iron oxides such as magnetite (Fe3O4) are the promising candidates for enzyme immobilization as offer high specific areas, low operational costs, facility of reuse and high enzyme loading capability.11–13,20,21 Furthermore, the catalyst can be recovered with an external magnet due to its magnetic properties, allowing the material recovery without need for tedious separation steps,22 also allowing less energy consumption and catalyst loss.21
Meanwhile, magnetic Fe3O4 particles tend to aggregate in liquid, which can be attributed to the strong magnetic dipole–dipole attractions among particles and there are not many active groups on the surface of Fe3O4 particles. Therefore, in order to affix the enzyme to the magnetic support a functionalization of the material should be conducted via the introduction of functional groups, which makes them suitable for catalysis applications.21,22
The concept of green chemistry is based in 12 principles including the atom economy, the use of reusable catalysts and the minimization of waste,23 which makes the development and use of magnetically recoverable biocatalyst even more relevant.
Engaged in the development of greener catalysts and their applications for environmental purposes, we proposed the synthesis of a magnetic biocatalyst prepared through the immobilization of soybean peroxidase onto the Fe3O4 particles. Magnetic Fe3O4–SiO2 particles were synthesized by coating with silicon to form a core–shell structure, and modified by silanization of (3-aminopropyl)triethoxysilane (APTES) to acquire NH2-functionalized Fe3O4–SiO2. Later, the immobilization of soybean peroxidase onto the NH2-modified magnetic silica particles was attempted using glutaraldehyde as cross linker agent. During covalent immobilization the bonds formed between the support and the enzyme must be strong enough for that process is efficient. When specific functional groups on the surface of carrier are absent, the carrier is subjected to a chemical modification,24 as performed in this work. The immobilization by covalent attachment to water-insoluble carriers via glutaraldehyde is one of the simplest and most gentle coupling methods in enzyme technology.25 Glutaraldehyde CH2(CH2CHO)2 is a bifunctional agent, needing –NH2 functions both in carrier and in protein, although it may eventually react with other groups (thiols, phenols, and imidazoles).26,27 The reactive aldehyde groups at the two ends of glutaraldehyde react with free amino groups of enzymes through a base reaction.17 The choice of glutaraldehyde as cross linker agent is due to its commercial availability, low cost and high reactivity. In addition, other advantage of method is the insertion of a long molecular spacer between support and protein, minimizing steric hindrance issues.26
The enzyme used in this work was extracted from soybean seed hulls, a soybean processing industry waste. The physicochemical properties of the material were determined and the obtained biocatalyst was appointed as a promising alternative for phenolic compound removal. The catalyst obtained, besides being recoverable and allowing its reuse, promotes waste minimization, since the peroxidase enzyme used was extracted from an agroindustrial waste. It is noteworthy that this is the first report wherein the soybean peroxidase was immobilized onto magnetic particles, resulting in magnetically separable catalysts, which can be an attractive alternative in the transformation of organic compounds, attending at least two of the 12 green chemistry principles (use of reusable catalysts and waste minimization).
Experimental
Enzyme formulation
The soybean peroxidase (SP) was extracted from soybean hulls, which are an agroindustrial waste generated in huge quantities in the soybean oil extraction process.28 The procedure for SP formulation was carried out according to Silva et al.29The peroxidase activity assay was based on the rate of guaiacol oxidation to tetraguaiacol (ε = 26.6 mM−1 cm−1). One unit of peroxidase activity at pH 6.0 represents the oxidation of 1 μmol of guaiacol per minute.30
Ferulic acid degradation by free peroxidase: determination of optimal reaction parameters
In order to determine the optimum reaction conditions for ferulic acid removal by the free enzyme, the degradation assays were carried out in glass reaction flasks at 30 °C, at a constant pH (citrate phosphate buffer 0.05 mol L−1; pH 6.0)29 by varying the following process parameters: reaction time, phenolic compound concentration (0.25–5.0 mmol L−1), enzyme activity (0.94–38 U mL−1) and H2O2 concentration (0.25–12 mmol L−1). Controls were carried out in the absence of H2O2. The reaction mixture was stirred continuously.The monitoring of ferulic acid degradation were carried out as follows: aliquots of the reaction mixture were taken at determined intervals up to 30 minutes, and the enzymatic reaction was stopped by adding 0.1 mL of catalase solution (1.2 mg of the commercial enzyme in 1.0 mL of 0.1 mol L−1, phosphate buffer, pH 7.0).9 The insoluble product was removed by centrifugation at 3000 × g, for 10 minutes at 25 °C. Ferulic acid residual concentration was measured by the colorimetric method of Folin and Denis using ferulic acid as standard.31
The optimum reaction conditions obtained for ferulic acid degradation by free peroxidase were used subsequently on the degradation assays with SP immobilized onto magnetic particles.
Preparation of Fe3O4–SiO2 particles modified
In a typical synthesis, an aqueous solution of Fe (SO4)2·7H2O (53 mmol, 20 g in 140 mL of distilled water) was placed in a 500 mL beaker. The solution was stirred at temperature of 90 °C and purged with N2. Next, 1.62 g of KNO3 (16 mmol) and 11.23 g of KOH (200 mmol) were added to 60 mL of distilled water and placed in a round bottom flask. Then this solution was added dropwise into the ferrous solution. The stirring was continued vigorously for 1 hour under N2 at 90 °C. The black precipitate generated was collected by magnetic separation, washed with water to neutral pH. The Fe3O4 particles were dried at 60 °C and kept in a desiccator.22A mixture of ethanol (200 mL) and distilled water (20 mL) was added to 2 g of magnetite particles, and the resulting dispersion was sonicated for 1 hour. Next, 5 mL of ammonia water and tetraethyl orthosilicate (TEOS, 6 mL) were added to the reaction solution. The resulting dispersion was under mechanically stirred continuously for 3 hours at room temperature. Next, to afford NH2-modified magnetic silica particles, 4 mL of APTES (3-aminopropyltriethoxysilane) were added, and the mixture was agitated for 12 hours. The NH2-modified magnetic silica particles were collected through vacuum filtration, followed by washing with water and ethanol, and then dried overnight in an oven at 60 °C.32
Immobilization of soybean peroxidase onto magnetic particles
Initially, 2 g from Fe3O4–SiO2–NH2 particles were dispersed in 100 mL of glutaraldehyde solution (1.6% v/v) at 25 °C for 3 hours. Then the carriers were washed with phosphate buffer (0.1 mol L−1, pH 7.0) and distilled water. Subsequently, the magnetic particles treated with GA were added into 20 mL of enzymatic solution (2 g of soybean peroxidase in 20 mL of citrate phosphate buffer 0.1 mol L−1, pH 6.0) and stirred continuously for 10 hours at 4 °C for the immobilization. Aliquots were taken at specific time intervals to determine the residual enzyme activity and the maximum immobilization yield.Immobilization yield (%) is defined as the difference obtained between the initial free enzyme activity of the peroxidase solution before the immobilization and the activity of free peroxidase obtained in the supernatant after immobilization divided by the enzyme activity before immobilization.
Characterization of materials
The crystalline structure of the materials were analysed by X-ray diffraction (XRD) (X'pert Pro X-ray diffractometer with Cu Kα radiation (λ = 0.15406 nm)). The crystallite size was estimated by X-ray powder diffraction patterns from measurement of the half-height width to the crystallographic (3 1 1) plane after correcting for instrumental broadening effects, using Scherrer equation.20 For corrections instrumental broadening was used with polycrystalline silicon standard. Transmission electron microcopy (TEM/HRTEM) images and energy dispersive analysis (EDS) of materials were obtained with a PHILIPS FEI TECNAI G20 F20 microscope operating at 200 kV. The Scanning Electronic Micrographs (SEM) were obtained using LEO 440 with an Oxford detector, the electron beam operating at 15 kV.Ferulic acid degradation by magnetic biocatalyst and reusability
The magnetic biocatalyst (600 mg, which corresponds to 5.25 U) was dispersed into 0.1 mol L−1 phosphate buffer, pH 6.5 (2.4 mL) and added to 3 mL of ferulic acid solution (1 mmol L−1). The reaction was started with addition of 0.8 mL from H2O2 (2 mmol L−1). The reaction mixture was kept under stirring for 30 minutes, at 30 °C. After the end of reaction, the biocatalyst was immediately recovered by magnetic separation, and the concentration of ferulic acid remaining in solution was determined by a spectrophotometric method.33For the reusability experiments, the sequential degradation assays were carried out as previously described. The catalyst was captured by a magnet and reused in each reaction cycle, and the remaining concentration of ferulic acid was measured.
Determination of enzyme kinetic parameters
Degradation assays using ferulic acid and hydrogen peroxide as substrates were carried out to determine the kinetic parameters Km and Vmax (Michaelis–Menten constant and reaction maximum rate, respectively) for the free enzyme and magnetic biocatalyst. Measurements were performed with different substrate concentrations. Km and Vmax were transformed to Lineweaver–Burk plots and values were calculated from the slopes and intercepts of the curves according to the following equation (eqn (1)).
 | (1) |
Results and discussion
Ferulic acid degradation by free peroxidase: determination of optimal reaction parameters
The catalyst/substrate contact time is an important factor which significantly influences the kinetics of an enzymatic reaction. The conversion percentages as a function of time have been represented (Fig. 1). | ||
| Fig. 1 Reaction progress on the ferulic acid removal by SP. Reaction conditions: enzyme load – SP 23.0 U mL−1; concentration of ferulic acid in the reaction medium: 1 mmol L−1 and 2 mmol L−1 of H2O2 at pH 6.0. | ||
According to Fig. 1, in only 2 minutes of reaction, more than half of the pollutant had been removed (56.97% ± 0.02). After, there was a slight decreasing reaction rate and the ferulic acid removal at 30 minutes of incubation was 70.77% (±0.02). The peroxidase-catalyzed reactions show fast reaction kinetics, making it an attractive alternative for bioremediation purposes.2
Since there was ferulic acid removal in the absence of H2O2 (approximately 10.0%), the authors suggest a slight adsorption of phenolic compound on the catalyst surface.
The concentration of substrate and co-substrate also significantly influences the enzymatic degradation rates. Hence, in order to examine the effect of these two parameters, the degradation assays were carried out by variation of both concentrations.
The peroxidase-catalyzed reactions in the process of pollutants biodegradation have been limited by the low stability of these enzymes in the presence of their natural substrate, hydrogen peroxide. In this case, the reaction needs to be designed in a way that maintains the stability of enzyme. An alternative is to couple one enzyme generating hydrogen peroxide “in situ” to the target enzyme resulting in a continuous supply of this reagent at low concentrations thus preventing enzyme inactivation.34 Furthermore, multiple H2O2 additions to reaction medium provide a low effective concentration of this substrate, avoiding the loss of enzyme activity.35 In this way, we evaluated the effect of both substrates (ferulic acid and H2O2) in the catalytic efficiency.
We observed that the maximal acid ferulic removal was obtained for a concentration of 1 mmol L−1 of substrate. Subsequent increase in phenolic compound concentration above 1 mmol L−1 resulted in decreased reaction rate. A peroxidase-catalyzed oxidation reaction generally requires both a substrate and co-substrate. The substrate is a reducing substrate, AH2, and the co-substrate is peroxide, which can be either H2O2 or an organic hydroperoxide (ping-pong mechanism).36 We investigated the effect of H2O2 concentration on the degradation of ferulic acid. An examination of Fig. 2 suggests that the co-substrate concentration which provides the maximal enzymatic performance (71.92% ± 0.48) is 2 mmol L−1. At peroxide concentrations above 2 mmol L−1 no significant pollutant removal efficiency was observed.
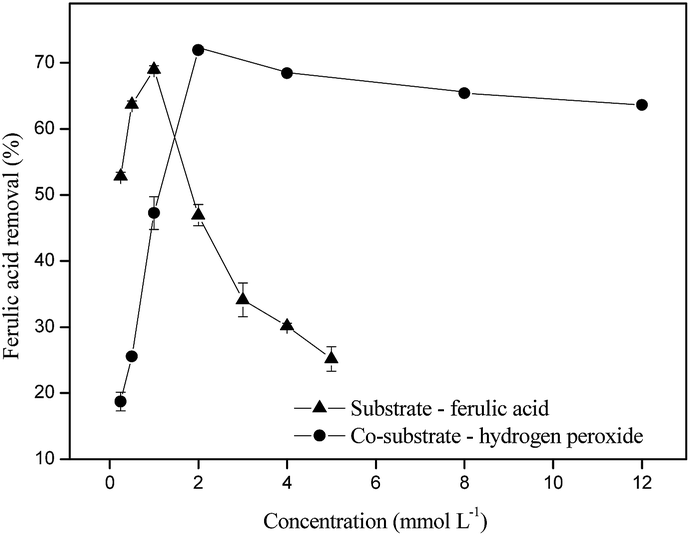 | ||
| Fig. 2 Effect of the concentration of substrate (ferulic acid) and co-substrate (H2O2) on the oxidation of ferulic acid by SP 23 U mL−1 at 30 minutes of reaction at pH 6.0. | ||
By gradually increasing of the catalyst concentration there was an increasing of acid ferulic removal until enzyme concentration of 23 U mL−1 (70.6%). The data presented in Fig. 3 suggest that the use of a saturated concentration of enzyme does not contribute to removal efficiency improvement, which may be attributed to a simultaneous decrease of substrate and co-substrate at elevated enzyme concentrations.37
 | ||
| Fig. 3 Effect of the concentration of enzyme on the oxidation of ferulic acid by SP 23 U mL−1 at 30 minutes of reaction. Reaction conditions: concentration of ferulic acid in the reaction medium: 1 mmol L−1 and 2 mmol L−1 of H2O2 at pH 6.0. | ||
Based upon the above data, we optimized the reaction parameters for the maximal pollutant removal. These parameters (substrate concentration 1 mmol L−1, H2O2 concentration 2 mmol L−1, enzyme load until 23 U mL−1 and time reaction 30 minutes) were utilized in the evaluation of efficiency of the previously prepared magnetic biocatalyst.
Preparation of magnetic biocatalyst
Magnetic particles Fe3O4–SiO2 were synthesized by coating with silica generated by hydrolysis and condensation of tetraethoxysilane (TEOS) and then modified by silanization of (3-aminopropyl)triethoxysilane (APTES) in order to make the reactive functional groups available. The silica matrix effectively prevents the clustering of the magnetic particles and provides a chemically inert surface for chemical modification.21 The functionalization or chemical modification of surface particles is required to anchor functional groups, such as amino groups, responsible for interactions between enzyme and support. Soybean peroxidase was then covalently immobilized on the surface of NH2-modified magnetic silica particles by using glutaraldehyde as a crosslinker.The time course of immobilization was investigated from 1 to 10 hours (Fig. 4). The approach used for monitoring the immobilization kinetics was to study the solution surrounding the particles through measurements in the supernatant after the protein particle suspension had been subjected to magnetic separation.
 | ||
| Fig. 4 The time course of the SP immobilization onto magnetic composites. | ||
According to the data obtained the highest yield of 89.23% was achieved after 1 hour of reaction. The yield reached a plateau, and there was no increase at 1 hour, only a slight decrease was observed at 2 hours of incubation. The immobilization occurs in ten minutes to hours, depending on particle concentration and protein type.38 The fast kinetics of the immobilization obtained in this work (only 1 hour) makes this biocatalyst attractive, since it showed an unusual bioimmobilization speed when compared to others magnetic biocatalysts described in the literature, where more than 10 hours were required for enzyme immobilization onto magnetic particles.10,32
Characterization of materials
The Fig. 5 shows the XRD patterns for magnetite nanoparticles, before and after surface modification. The all materials showed diffraction peaks at 2θ = 30.2°, 35.5°, 43.3°, 53.8°, 57.2° and 62.7° which are attributed to the (220), (311), (400), (422), (511) and (440) planes attributed to the fcc spinel phase of Fe3O4 (JCPDS no. 89-4319). | ||
| Fig. 5 XRD patterns of Fe3O4, Fe3O4–SiO2 core–shell nanoparticles modified with amino groups and final biocatalyst. | ||
The surface amino modification process of the silica coated magnetite did not result in the Fe3O4 phase change.
In addition, the average diameter of the crystallites were estimated according to Scherrer's equation. It should be noted that heating in a water bath for 3 hours during the silanization reaction did not significantly changed the magnetite particles growth. The average crystallite diameters were: bare Fe3O4 (33 nm), Fe3O4–SiO2 core–shell modified with amino groups (33 nm) and final biocatalyst (35 nm).
The morphological characteristics of all the materials were analysed by SEM and TEM (Fig. 6). The SEM images of bare Fe3O4 and final biocatalyst (Fe3O4–SiO2–NH2-peroxidase) are shown in Fig. 6A and B, respectively. SEM images of the final biocatalyst (Fig. 6B), revealed a significant change on surface of the material compared to the bare Fe3O4. The final biocatalyst showed particles with quasi-spherical morphology, suggesting that peroxidase was successfully covalently immobilized onto the surface of nanoparticles. Silica acts as a capping agent for each single cubic magnetite particle,38 which is in good agreement with the images obtained by TEM and HRTEM (Fig. 6C). In addition, an elemental analysis scan performed in scanning transmission electron mode (STEM) is shown for the final biocatalyst in Fig. 6E. The red line highlighted on the particle surface indicates where the EDX line scan spectrum was performed. The EDX line scan spectrum shows that the environment around the Fe3O4 particles consists of silicon (Si). However, on particle surface of Fe304, the Fe is the principal element, with a small amount of Si, confirming that silica acts as a capping agent.
 | ||
| Fig. 6 SEM images of bare Fe3O4 nanoparticles (A) and Fe3O4–SiO2–NH2-peroxidase (B); TEM images of naked Fe3O4 nanoparticles (C) and Fe3O4–SiO2 core–shell nanoparticles modified with amino groups (D); scanning transmission electron mode (STEM) of Fe3O4–SiO2–NH2-peroxidase (E). | ||
It is important to mention that, compared with bare Fe3O4 nanoparticles, the magnetic particle morphology and size, after chemical modification, were not changed, due the reaction occurring only on the particle surface.10 The DRX data also corroborate with the data taken from the TEM analysis.
Ferulic acid degradation by magnetic biocatalyst and reusability
The ferulic acid degradation by hybrid peroxidase catalyst was investigated. The optimal parameters obtained for free enzyme were utilized in the degradation assays (substrate concentration 1 mmol L−1, H2O2 concentration 2 mmol L−1 and time of reaction 30 minutes). The mass of biocatalyst used was 600 mg, which corresponds to enzyme activity of 5.25 U. The ferulic removal was carried out using both free and immobilized enzyme at 5.25 U at the same reaction conditions. The ferulic acid removal obtained was 57.67 ± 0.27% and 99.67 ± 0.10% for free and immobilized enzyme, respectively. Therefore, the resulting biocatalyst exhibited a remarkable catalytic efficacy in the phenol remediation.The reusability of the catalyst is an important feature and is critical for their application in industry or for environmental purposes. Thus, the reusability of immobilized SP was investigated in the ferulic acid degradation under the same reaction conditions previously used. After the completion of the reaction, in the first cycle, the catalyst was recovered from the reaction solution by applying a magnetic field, washed with citrate phosphate buffer (0.05 mol L−1) pH 6.0 and water to remove any remaining substrate and product before the next experiment. As shown in Fig. 7, there was a pollutant removal loss from 76.3% to 47.46% in the second cycle.
 | ||
| Fig. 7 Reuse performance of catalyst after each cycle for six cycles. Reaction conditions: enzyme load – 600 mg; concentration of ferulic acid in the reaction medium: 1 mmol L−1 and 2 mmol L−1 of H2O2 at pH 6.0. | ||
Next, the rate of phenolic removal remained constant until the fifth cycle indicating that the immobilized enzyme has good reusability and can be regenerated for repeated use. Furthermore the reuse of the immobilized enzyme is very easy due to the superparamagnetism of the magnetic support. The decrease performance of the catalyst after each cycle may be attributed to the accumulation of the end-product polymer (formed during the oxidative polymerization of phenolic compounds by peroxidases) on the catalyst surface hindering the access of the substrate to the enzyme active site.9 Several authors suggest that the decrease in enzymatic activity after successive cycles is related to some enzyme leaching from the support.15,39 Therefore, the amount of enzyme loss during the each cycle was measured. However, from the first cycle on, no catalyst loss was detected, implying a strong bonding of SP on the support.
Determination of enzyme kinetic parameters
Accepting that enzymes operate mostly under steady-state conditions, the analyses of Michaelis kinetics parameters are more beneficial for the understanding of the catalytic process. The Michaelis constant (Km) is an equilibrium constant estimate for substrate binding to the enzyme. The lower Km value represents higher affinity between enzymes and substrates.29,40,41The kinetic parameters of free and immobilized peroxidase are listed in Table 1 for both substrates.
| Km (mM) | Vmax (mM min−1) | Vmax/Km (min−1) | |
|---|---|---|---|
| Ferulic acid as substrate | |||
| Free enzyme | 607.03 | 6.21 | 0.01 |
| Immobilized enzyme | 21.55 | 0.654 | 0.03 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|||
| Hydrogen peroxide as substrate | |||
| Free enzyme | 0.075 | 0.009 | 0.12 |
| Immobilized enzyme | 0.200 | 0.035 | 0.075 |
As illustrated in Table 1, Km value of the immobilized enzyme for ferulic acid is 28-fold lower (21.55 mmol L−1) than the Km value of the free enzyme (607.33) indicating a higher affinity of the magnetic biocatalyst toward the pollutant. This result can be explained by the positive effect of immobilization in terms of reduced steric hindrance or substrate diffusion resistance.42 However, concerning to hydrogen peroxide, a increase of Km value was observed to immobilized enzyme. The Km value is dependent on the characteristics of both enzyme and substrate,43 therefore, the kinetic parameters determined for hydrogen peroxide showed an unlike behaviour. Several authors suggest that after the immobilization an increasing of Km value may occur, which can be attributed to the lower possibility to form the substrate–enzyme complex caused by the conformation changes of the enzyme.44–50 In this present work, the affinity of peroxidase for ferulic acid, a model pollutant, increased after immobilization, which makes the catalyst obtained a promising alternative. The Vmax/Km ratio of free and immobilized enzyme was 0.01 and 0.03 min−1, respectively. The higher the Vmax/Km value, the higher the catalytic efficiency of the immobilized enzyme for this pollutant. Therefore, according to the kinetic parameters obtained, we concluded that the catalyst developed exhibited higher bioremediation efficiency compared with the free enzyme. Regarding to H2O2 substrate, the increase in Km of immobilized enzyme indicated that immobilized enzyme has an apparent lower affinity for this substrate.
Conclusions
The use of nanobiocatalysts is drawing great attention as an innovative technology. In this work, a recoverable magnetic biocatalyst was prepared via immobilization of soybean peroxidase onto magnetite nanoparticles modified with amino groups. The high immobilization yield (above 89%) was obtained in only 1 hour of reaction. In contrast to conventional use of free enzymes, the catalyst produced showed the highest catalytic efficiency and good reusability making them more attractive for industrial applications. The prepared catalysts have a strong magnetic responsivity for separation under an applied magnetic field and showed potential in the acid ferulic removal, a model pollutant. On the basis of these results, we concluded that the prepared magnetic nanoparticles can be considered a high-performance nanocatalyst for environmental remediation.Acknowledgements
The authors thank the Coordenadoria de Aperfeiçoamento de Pessoal de Nível Superior (Capes – Brazil) and Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq-Brazil) for the scholarships, the Universidade Federal de Lavras and Universidade Federal de Minas Gerais for the technical support.References
- M. A. Rao, R. Scelza, F. Acevedo, M. C. Diez and L. B. Gianfreda, Chemosphere, 2014, 35, 145 CrossRef PubMed.
- J. A. Torres, P. M. B. Chagas, M. C. Silva, C. D. dos Santos and A. D. Corrêa, Water Sci. Technol., 2016, 73, 39 CrossRef PubMed.
- X. Duan, S. C. Corgié, D. J. Aneshansley, P. Wang, L. P. Walker and E. P. Gianneĺs, ChemPhysChem, 2014, 15, 974 CrossRef CAS PubMed.
- J. Huang, Q. Chang, Y. Ding, X. Han and H. Tang, Chem. Eng. J., 2014, 15, 434 CrossRef.
- L. Hong-Mei and J. A. Nicell, Bioresour. Technol., 2008, 99, 4428 CrossRef PubMed.
- Y. Jiang, C. Cui, L. Zhou, Y. He and J. Gao, Ind. Eng. Chem. Res., 2014, 53, 7591 CrossRef CAS.
- X. G. Xie and C. C. Dai, Bioresour. Technol., 2015, 179, 35 CrossRef CAS PubMed.
- B. R. Yadav and A. Garg, Chem. Eng. J., 2014, 252, 185 CrossRef CAS.
- J. A. Torres, P. M. B. Chagas, M. C. Silva, C. D. dos Santos and A. D. Corrêa, Environ. Technol., 2015, 23, 1 Search PubMed.
- Y. Li, X. Y. Wang, X. P. Jiang, J. J. Ye, Y. W. Zhang and X. Y. Zhang, J. Nanopart. Res., 2015, 17, 8 CrossRef.
- X. Y. Wang, X. P. Jiang, Y. Li, S. Zeng and Y. W. Zhang, Int. J. Biol. Macromol., 2015, 75, 44 CrossRef CAS PubMed.
- M. Hartmann and X. Kostrov, Chem. Soc. Rev., 2013, 42, 6277 RSC.
- S. Datta, L. R. Christena and Y. R. S. Rajaram, Biotechnology, 2013, 3, 1 Search PubMed.
- N. R. Mohamad, N. H. C. Maarzuki, N. A. Buang, F. Huyop and R. A. Wahab, Biotechnol. Biotechnol. Equip., 2015, 29, 205 CrossRef CAS PubMed.
- Y. Cao, L. Wen, F. Svec, T. Tan and Y. Lv, Chem. Eng. J., 2016, 286, 272 CrossRef CAS.
- J. Xu, J. Sun, Y. Wang, J. Sheng, F. Wang and M. Sun, Molecules, 2014, 19, 11465 CrossRef PubMed.
- R. Ahmad and M. Sardar, Biochem. Anal. Biochem., 2015, 4, 178 Search PubMed.
- M. N. Gupta, M. Kaloti, M. Kapoor and K. Solanki, Artif. Cells, Blood Substitutes, Immobilization Biotechnol., 2011, 39, 98 CrossRef CAS PubMed.
- H. Vaghari, H. Jafarizadeh-Malmiri, M. Mohammadlou, A. Berenjian, N. Anarjan, N. Jafari and S. Nasiri, Biotechnol. Lett., 2016, 38, 223 CrossRef CAS PubMed.
- L. Shi, F. Ma, Y. Han, X. Zhang and H. Yu, J. Hazard. Mater., 2014, 279, 203 CrossRef CAS PubMed.
- R. K. Sharma, S. Dutta and S. Sharma, Dalton Trans., 2015, 44, 1303 RSC.
- Q. Zhang, X. Han and B. Tang, RSC Adv., 2013, 3, 9924 RSC.
- I. Fechete, Y. Wang and J. C. Védrine, Catal. Today, 2012, 2, 27 Search PubMed.
- T. Jesionowski, J. Zdarta and B. Krajewska, Adsorption, 2014, 20, 801 CrossRef CAS.
- I. Migneault, C. Dartiguenave, M. J. Bertrand and K. C. Waldron, BioTechniques, 2004, 37, 790 CAS.
- P. Zucca and E. Sanjust, Molecules, 2014, 19, 14139 CrossRef PubMed.
- Y. Wine, N. Cohen-Hadar, A. Freeman and F. Frolow, Biotechnol. Bioeng., 2007, 98, 711–718 CrossRef CAS PubMed.
- W. P. F. Neto, H. A. Silvério, N. O. Dantas and D. Pasquini, Ind. Crops Prod., 2013, 42, 480 CrossRef.
- M. C. Silva, J. A. Torres, A. A. Castro, E. F. F. da Cunha, L. C. A. de Oliveira, A. D. Corrêa and T. C. Ramalho, J. Biomol. Struct. Dyn., 2015, 34, 1839 Search PubMed.
- A. A. Khan and D. S. Robinson, Food Chem., 1994, 49, 407 CrossRef CAS.
- Association of Official Analytical Chemists, Official methods of analysis, Pharmabooks, Gaithersburg, 19th edn, 2012 Search PubMed.
- Q. Chang and H. Tang, Molecules, 2014, 19, 15768 CrossRef CAS PubMed.
- H. Klung and L. Alexander, X-ray diffraction procedures, Wiley, New York, 1962, p. 491 Search PubMed.
- K. Hernandez, A. Berenguer-Murcia, R. C. Rodrigues and R. Fernandez-Lafuente, Curr. Org. Chem., 2012, 16, 2652 CrossRef CAS.
- A. Heinfling-Weidtmann, T. Reemtsma, T. Storm and U. Szewzyk, FEMS Microbiol. Lett., 2011, 203, 179 CrossRef.
- J. Zhao, C. Lu and S. Franzen, J. Phys. Chem. B, 2015, 119, 12828 CrossRef CAS PubMed.
- M. C. Silva, J. A. Torres, L. R. V. Sá, P. M. B. Chagas, V. S. Ferreira-Leitão and A. D. Corrêa, J. Mol. Catal. B: Enzym., 2013, 89, 122 CrossRef CAS.
- P. S. N. Zadeh, K. A. Mallak, N. Carlsson and B. Åkerman, Anal. Biochem., 2015, 476, 51 CrossRef PubMed.
- R. Roto, Y. Yusran and A. Kuncaka, Appl. Surf. Sci., 2016, 377, 30 CrossRef CAS; K. Ramani, S. Karthikeyan, R. Boopathy, L. John Kennedy, A. B. Mandal and G. Sekaran, Process Biochem., 2012, 47, 435 CrossRef.
- J. M. Lee, G. R. Xu, B. K. Kim, H. N. Choi and W. Y. Lee, Electroanalysis, 2011, 23, 962 CrossRef CAS.
- N. Sohrabi, N. Rasouli and M. Torkzadeh, Chem. Eng. J., 2014, 240, 426 CrossRef CAS.
- J. Long, X. Li, Z. Wu, E. Xu, X. Xu, Z. Jin and A. Jiao, Colloids Surf., A, 2015, 472, 69 CrossRef CAS.
- V. Kumar, N. Misra, N. K. Goel, R. Thakar, J. Gupta and L. Varshney, RSC Adv., 2016, 2974 RSC.
- A. P. M. Tavares, C. G. Silva, G. Drazic, A. M. T. Silva, J. M. Loureiro and J. L. Faria, J. Colloid Interface Sci., 2015, 454, 52 CrossRef CAS PubMed.
- M. Deng, H. Zhao, S. Zhang, C. Tian, D. Zhang, P. Du, C. Liu, H. Cao and H. Li, J. Mol. Catal. B: Enzym., 2015, 112, 15 CrossRef CAS.
- G. Bayramoglu and M. Y. Arıca, J. Mol. Catal. B: Enzym., 2008, 55, 83 CrossRef.
- F. Secundo, Chem. Soc. Rev., 2013, 42, 6250 RSC.
- U. Guzik, K. Hupert-Kocurek and D. Wojcieszyńska, Molecules, 2014, 19, 8995 CrossRef PubMed.
- R. C. Rodrigues, C. Ortiz, A. Berenguer-Murcia, R. Torres and R. Fernández-Lafuente, Chem. Soc. Rev., 2013, 42, 6290 RSC.
- M. L. Verma, M. Naebe, C. J. Barrow and C. J. Puri, PLoS One, 2013, 8, e73642 CAS.
| This journal is © The Royal Society of Chemistry 2016 |