
Grafting modification of epoxidized natural rubber with poly(ethylene glycol) monomethylether carboxylic acid and ionic conductivity of graft polymer composite electrolytes
Abstract
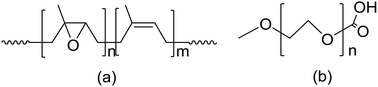
A novel comb-like polymer was synthesized via grafting epoxidized natural rubber (ENR) with polyethylene glycol (PEG) monomethylether carboxylic acid (mPEG-COOH), and the grafting reaction was studied by variable-temperature Fourier transform infrared (FTIR) spectroscopy. This comb-like polymer was characterized by attenuated total reflectance-FTIR (ATR-FTIR) spectroscopy and differential scanning calorimetry (DSC). The carboxyl groups of mPEG-COOH reacted with the epoxy groups of ENR to produce graft polymers. The composite polymer electrolyte (CPE) based on this comb-like polymer ENR-g-mPEG-COOH was prepared via introducing LiClO4 into the graft polymer matrix. The CPE was studied by X-ray diffraction analysis (XRD), DSC, an equilibrium swelling method, and electrochemical workstation techniques. With increasing mPEG-COOH content, the ionic conductivity of the electrolyte significantly increased.
 Please wait while we load your content...
Please wait while we load your content...

