
Competent overall water-splitting electrocatalysts derived from ZIF-67 grown on carbon cloth†
Xin Liuab,
Jinmei Dongab,
Bo Youa and
Yujie Sun*a
aDepartment of Chemistry and Biochemistry, Utah State University, Logan, Utah 84322, USA. E-mail: yujie.sun@usu.edu; Fax: +1-435-797-3390; Tel: +1-435-797-7608
bKey Laboratory of Comprehensive and Highly Efficient Utilization of Salt Lake Resources, Chinese Academy of Sciences, 18 Xinning Road, Xi'ning 810008, China
First published on 26th July 2016
Abstract
The design of nonprecious, bifunctional, and highly competent electrocatalysts for both H2 and O2 evolution reactions (HER and OER) has attracted increasing interest recently. Herein, we report a cobalt-based electrocatalyst derived from ZIF-67 grown on carbon cloth (Co–P/NC/CC) for overall water splitting electrocatalysis. The as-prepared Co–P/NC/CC catalyst exhibited remarkable catalytic performance in 1 M KOH with Tafel slopes of 52 and 61 mV dec−1 for HER and OER, respectively. When serving as catalysts for both the cathode and anode, our Co–P/NC/CC demonstrated high efficiency and strong robustness. A thorough comparison with other control samples and detailed characterization results revealed that the superior activity and excellent stability of Co–N–C likely originated from the highly porous, self-supported, and binder-free nature of the electrocatalyst, as well as the high conductivity of carbon cloth. Hence, direct decoration of metal organic frameworks on conductive substrates represents an effective approach for the development of electrocatalysts not only promising for water splitting but also for many other applications.
1. Introduction
Along with the increasing global energy demands and deteriorating climate change resulting from fossil fuel utilization, clean and renewable energy-driven water splitting to produce H2 and O2 has been widely considered as a promising approach to alleviate our reliance on fossil fuels and mitigate climate change.1,2 The slow kinetics of both H2 and O2 evolution reactions (HER and OER) necessitate the development of competent electrocatalysts.3,4 Even though platinum, iridium- and ruthenium-based oxides have long been recognized as start-of-the-art electrocatalysts for HER and OER, respectively, their low abundance and thus high cost call for the exploration of alternative earth-abundant catalyst candidates. Indeed, recent years have witnessed the emergence of many excellent HER and OER catalysts composed of low-cost materials, such as 1st-row transition metals,5 oxides,6 oxyhydroxides,7–9 sulfides,10–14 selenides,15,16 phosphides,17–19 nitrides,20 as well as metal-free electrocatalysts.21 The large earth abundance and rich redox properties of these catalysts make them quite promising for large scale employment of electrocatalytic water splitting.22,23 In order to accomplish high energy conversion efficiency, water splitting is better to be conducted in either strongly acidic or alkaline solution to minimize the electrolyte resistance.24,25 Therefore, it requires that the HER and OER catalysts are able to function in the same medium.26 Within this context, it might be cost competitive to develop bifunctional electrocatalysts (or catalyst precursors) which are active for both HER and OER in the same electrolyte.Taking advantage of the large specific surface area and high porosity of metal organic frameworks (MOF), our group recently reported several MOF-derived electrocatalysts for various reactions, such as water splitting and oxygen reduction reaction.27,28 We demonstrated that a cobalt imidazolate-based MOF, ZIF-67, was able to act as a bifunctional electrocatalyst for overall water splitting after pyrolysis and phosphidation treatments.27 In order to load the ZIF-67-derived catalyst on the working electrode, a conductive polymer, Nafion, was utilized to immobilize the catalyst on the electrode surface. Such a viscous binder of Nafion will inevitably result in buried active sites, decreased specific surface area, and increased interfacial resistance, which would result in inferior performance of the catalyst and complicate the electrode preparation step.29–31 Therefore, we seek to develop MOF-derived electrocatalysts directly grown on conductive substrates with intimate physical and electric contact.32 To the best of our knowledge, direct growing of MOF-derived electrocatalysts on conductive substrates for overall water splitting has received scarce attention.
Herein, we report a facile and effective strategy to anchor ZIF-67 on the surface of carbon cloth (CC, Fig. S1a†) to yield ZIF-67/CC. Carbon cloth is a type of cheap and commercially available textile with high conductivity, robust corrosion resistance, excellent flexibility, and strong mechanical strength, representing an ideal 3D substrate to integrate catalysts for electrocatalytic applications.33–36 Pyrolysis of ZIF-67/CC followed by phosphidation resulted in cobalt phosphide nanoparticles imbedded in nitrogen-containing carbon matrices anchored on carbon cloth tightly (Co–P/NC/CC). Electrochemical studies of Co–P/NC/CC demonstrate its excellent activities for electrocatalytic HER, OER, and overall water splitting. A myriad of materials characterization techniques were also employed to study the composition and morphology of Co–P/NC/CC prior to and post electrocatalytic reactions. The excellent electrocatalytic performance of Co–P/NC/CC compared to control samples, such as ZIF-67-derived catalyst particles drop casted on carbon cloth, is primarily attributed to the intimate contact between Co–P/NC catalyst and carbon cloth, as well as the large surface area and high conductivity of the latter.
2. Experimental
2.1 Chemicals
2-Methylimidazole, potassium hydroxide and sodium hypophosphite monohydrate were purchased from Alfa Aesar. Cobalt nitrate hexahydrate and Nafion 117 solution (5% in a mixture of lower aliphatic alcohols and water) were purchased from Sigma Aldrich. Methanol was purchased from Decon Laboratories. All chemicals were used as received without any further purification. Carbon cloth was purchased from Fuel Cell Store. Deionized water (18 MΩ cm) obtained from a Barnstead E-Pure system was used in all experiments.2.2 Syntheses
2.3 Physical methods
Scanning electron microscopy (SEM) and elemental mapping analysis were conducted on a FEI QUANTA FEG 650 (FEI, USA). X-ray diffraction (XRD) data were recorded on a Rigaku MinifexII Desktop X-ray diffractometer. The cobalt and phosphorus quantities of each catalyst sample were analysed by a Thermo Electron iCAP inductively coupled plasma atomic emission spectrophotometer (ICP-AES). X-ray photoelectron spectra were collected on a Kratos Axis Ultra instrument (Chestnut Ridge, NY). The catalyst samples were affixed on a stainless steel Kratos sample bar, loaded into the instrument's load lock chamber, and evacuated to 5 × 10−8 Torr before they were transferred into the sample analysis chamber under ultrahigh vacuum conditions (∼10−10 Torr). X-ray photoelectron spectra were taken using the monochromatic Al Kα source (1486.7 eV) at a 300 × 700 μm spot size. High resolution regions at the binding energy of interest were taken for each sample. The samples were also sputter cleaned inside the analysis chamber with 1 keV Ar+ ions for 30 seconds to remove adventitious contaminants. The XPS data were analysed using CASA XPS software and energy corrections on high resolution scans were calibrated by referencing the C 1s peak of adventitious carbon to 284.5 eV.2.4 Electrocatalytic measurements
Linear sweep voltammetry (LSV) was performed by a computer-controlled Gamry Interface 1000 electrochemical workstation with a three-electrode configuration. The catalyst-coated carbon cloth was directly used as the working electrode. A Ag/AgCl (sat. KCl) electrode was used as the reference electrode and a Pt wire as the counter electrode. All potentials reported herein were referenced to the reversible hydrogen electrode (RHE) through calibration. 1.0 M KOH was utilized. Electric impedance spectroscopy was carried out in the same configuration from 105 to 0.1 Hz with an AC potential amplitude of 30 mV. The electrochemical double-layer capacitance (Cdl) of each sample was evaluated via cyclic voltammetry in a non-faradaic region from 0.123 to 0.223 V vs. RHE for HER and 1.223 to 1.323 V vs. RHE for OER at scan rates ranging from 4 to 40 mV s−1. The resulting linear slope is twice of the Cdl. For overall water splitting tests, the same-type catalyst-coated carbon cloth electrodes were used for anode and cathode directly. The final chronopotentiometry was conducted at a current density of 10 mA cm−2 for 20 h.3. Results and discussion
The overall synthetic route is illustrated in Scheme 1. In the presence of carbon cloth, a mixture of cobalt nitrate and 2-methylimidazole in methanol resulted in ZIF-67 preferentially grown on this conductive substrate, probably due to ample nucleation sites on the surface of carbon cloth. The as-prepared ZIF-67 anchored on carbon cloth was named as ZIF-67/CC. Fig. 1a and b presents the SEM images of ZIF-67, showing regular nano-polyhedrons grown on carbon cloth tightly. The size of these ZIF-67 polyhedrons is around 0.5–1 μm, close to reported samples.27 Element mapping analysis (Fig. 1c) suggested that ZIF-67 mainly consisted of Co, C, and N, all of which were homogeneously distributed throughout the entire nano-polyhedrons. | ||
| Scheme 1 Synthetic procedure of Co–P/NC/CC. | ||
 | ||
| Fig. 1 (a and b) SEM images of ZIF-67/CC and (c) the elemental mapping images of ZIF-67/CC showing the distributions of Co, C, and N. (d and e) SEM images of Co–P/NC/CC and (f) the elemental mapping images of Co–P/NC/CC showing the distributions of Co, P, C, and N. | ||
Subsequently, ZIF-67/CC was carburized under argon at 900 °C for 3 h. During this process, ZIF-67 nano-polyhedrons transformed to metallic cobalt nanoparticles imbedded in nitrogen-containing carbon matrices, which was denoted as Co/NC/CC. The SEM image of Co/NC/CC (Fig. S1b†) indicated that the original regular and smooth nano-polyhedrons of ZIF-67 shrank to rough porous composites. Nevertheless, the discrete nanopolyhedrons could still be identified. Next, Co/NC/CC was phosphatized under argon at 300 °C for 2 h, utilizing sodium hypophosphite as the phosphorous source and resulting in cobalt phosphide nanoparticles decorated within nitrogen-containing carbon matrices. This final product was named as Co–P/NC/CC. As shown in Fig. 1d, the SEM image of Co–P/NC/CC proved that the integrity of each shrunk nano-polyhedron was preserved, although a close inspection (Fig. 1e) implied some agglomeration. It should be noted that these new nano-polyhedrons were still tightly anchored on the surface of carbon cloth. The large enrichment of phosphorous in these nano-polyhedrons was well manifested in the elemental mapping images shown in Fig. 1f, along with the homogeneous distribution of Co, P, N, and C.
The XRD pattern of ZIF-67/CC (Fig. S2†) confirmed the crystallinity and identity of ZIF-67 grown on carbon cloth.37,38 After pyrolysis, the weak and broad XRD peaks of Co/NC/CC (Fig. S3†) at 44.2° and 51.5° suggested the formation of metallic cobalt; while the relatively intense peak around 25–30° was due to the substrate of carbon cloth. After phosphidation, despite the lack of well-resolved XRD peaks for Co–P/NC/CC, a comparison with standard XRD patterns of CoP and Co2P suggested the formation of a mixture of CoP and Co2P in Co–P/NC/CC (Fig. S4†). Elemental analysis data listed in Table S1† presented a Co/P atomic ratio is close to 4.5 (much larger than 2), implying that only a layer of CoP and Co2P mixture was formed on the surface while the inner composite was still metallic cobalt in Co–P/NC/CC.
In order to further shed light on the composition and valence states of the elements in each catalyst sample, X-ray photoelectron spectroscopy (XPS) was collected. Fig. S5a† presents the survey spectrum of Co–P/NC/CC which showed all the anticipated elements of Co, P, C, and N, consistent with the elemental mapping results (Fig. 1f). The high-resolution Co 2p XPS spectrum of Co–P/NC/CC is plotted in Fig. 2a. Deconvolution of this Co 2p spectrum resulted in primary Co 2p3/2 peaks at 778.5 and 782.1 eV, corresponding to cobalt phosphides and cobalt oxides, respectively. The presence of cobalt oxides was probably due to oxidation in air prior to the XPS measurement.33,39 The weak feature around 793.6 eV could be assigned to the Co 2p1/2 binding energy. The deconvolution of the high-resolution P 2p XPS spectrum led to a prominent peak at 134.0 eV, attributable to oxidized phosphorous species (e.g., phosphate). In the meantime, typical phosphide signal could still be observed at 129.7 and 131.1 eV, which were attributed to the P 2p3/2 and 2p1/2 features of phosphorous in cobalt phosphide. Overall, the binding energies at 778.5 and 129.7 eV were quite close to those of CoP.40 In addition, the high-resolution N 2p XPS spectrum was also collected and deconvoluted in Fig. S5b,† displaying binding energies at approximately 398.9, 399.4, and 401.7 eV, which could be attributed to pyridinic N, Co–N, and graphitic N, respectively.
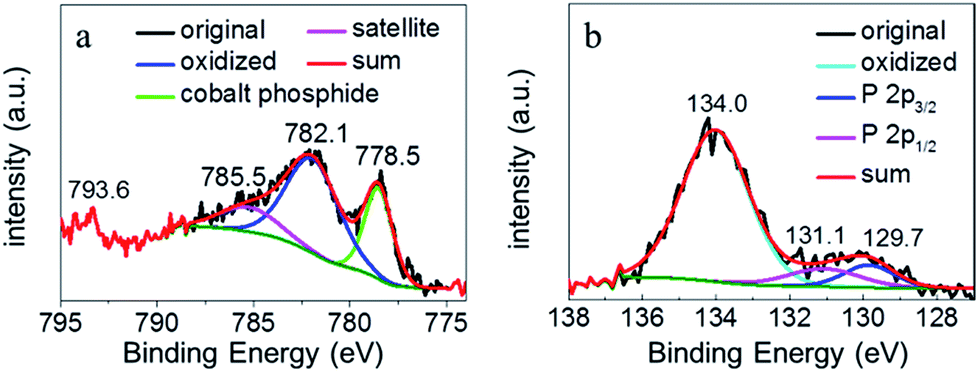 | ||
| Fig. 2 High-resolution of XPS spectra of Co–P/NC/CC in the (a) Co 2p region and (b) P 2p region. | ||
With the detailed characterization data in hand, we next assessed the electrocatalytic performance of these catalysts for H2 evolution. All the electrochemical experiments were conducted in 1.0 M KOH using a three-electrode configuration unless otherwise noted. The linear sweep voltammetry (LSV) curves of Co–P/NC/CC and control samples (carbon cloth, ZIF-67/CC, and Co/NC/CC) were plotted in Fig. 3a. It is apparent that Co–P/NC/CC catalyzed H2 evolution much earlier than all the control samples with a catalytic onset potential around −0.1 V vs. reversible hydrogen electrode (RHE). Without phosphidation, Co/NC/CC initiated HER catalytic current approximately 50 mV more negative than that of Co–P/NC/CC. Most likely due to the poor conductivity and lack of active sites in ZIF-67, ZIF-67/CC only acted slightly better than the background carbon cloth for HER. Specifically, Co–P/NC/CC only required overpotentials of −171 and −243 mV to reach catalytic current densities of 10 and 100 mA cm−2, respectively. These overpotential requirements are much smaller than those of Co/NC/CC and ZIF-67/CC to achieve the same current densities and also compare quite favourably with those of recently reported HER electrocatalysts in strongly alkaline electrolytes (see detailed comparison in Table S2†).41–44 Even though Pt/C exhibited a smaller onset potential, the current density of our Co–P/NC/CC surpassed that of Pt/C beyond −220 mV vs. RHE (Fig. S8a†). Fig. 3b presented the derived Tafel plots of each catalyst and apparently Co–P/NC/CC exhibited a much smaller Tafel slope (52 mV dec−1) compared to those of carbon cloth (81 mV dec−1), ZIF-67/CC (75 mV dec−1), and Co/NC/CC (100 mV dec−1), implying a faster HER kinetic rate of the former. In addition, electric impedance spectroscopy was collected to further probe the superior HER activity of Co–P/NC/CC. As shown in Fig. 3c, the Nyquist plot of Co–P/NC/CC showed a smaller semi-circular diameter than those of the control samples and thus demonstrated a lower resistance for electrocatalytic HER. In order to determine whether the high HER performance of Co–P/NC/CC is due to its increased electrochemically active surface area (ECSA), we next measured its double-layer capacitance via cyclic voltammetry in the non-faradaic region (0.123 to 0.223 V vs. RHE, Fig. S6†). It has been widely accepted that cyclic voltammetry at non-faradaic potentials is able to estimate ECSA, especially when the electrodes consist of similar composites. By plotting the difference in current density between the anodic and cathodic scans at 0.173 V vs. RHE, nearly linear plots could be obtained in Fig. 3d. The extracted slopes of the linear fitting resulted in similar ECSA for both Co–P/NC/CC and Co/NC/CC, which were substantially larger than those of ZIF-67/CC and carbon cloth. Therefore, it is clear that although large ECSA is beneficial towards HER, the higher HER activity of Co–P/NC/CC relative to Co/NC/CC is most likely owning to the phosphidation step which yielded more HER active cobalt phosphides on the cobalt nanoparticles.
 | ||
| Fig. 3 (a) LSV curves of Co–P/NC/CC (purple), Co/NC/CC (blue), ZIF-67/CC (red), and CC (black) for HER in 1.0 M KOH at a scan rate of 2 mV s−1. (b) Tafel plots of Co–P/NC/CC (purple), Co/NC/CC (blue), ZIF-67/CC (red), and CC (black) for HER. (c) Nyquist plots of Co–P/NC/CC (purple), Co/NC/CC (blue), ZIF-67/CC (red), and CC (black) measured at −247 mV vs. RHE. Inset shows the expanded region around the intercepts at the x axis. (d) Scan rate dependence of the current densities of Co–P/NC/CC (purple), Co/NC/CC (blue), ZIF-67/CC (red), and CC (black) at 0.173 V vs. RHE. | ||
The OER performances of these catalysts were also evaluated in the same electrolyte, 1.0 M KOH. Apparently, Co–P/NC/CC exhibited a much smaller onset potential of ∼1.5 V vs. RHE and greater catalytic current than those of Co/NC/CC, ZIF-67/CC, and CC (Fig. 4a). Noticeably, Co/NC/CC required an overpotential of 360 mV to reach a current density of 10 mA cm−2; while after phosphidation, Co–P/NC/CC only required an overpotential of 330 mV to arrive at the same current density. ZIF-67/CC and carbon cloth were not very active towards OER and they required potential more positive than 1.6 V to produce appreciable catalytic current. Even though these catalysts exhibited substantially different catalytic onsets, their derived Tafel slopes fell in a close range of 50–70 mV dec−1. Electric impedance spectra of these catalysts are shown in Fig. 4c, wherein Co–P/NC/CC presented the smallest resistance, in agreement with its best OER activity. Despite the similarity in the electrochemical surface areas of Co–P/NC/CC and Co/NC/CC for HER application, a rather obvious enhancement in ECSA was observed for Co–P/NC/CC relative to Co/NC/CC for O2 evolution (Fig. 4d and S7†). It implied that during electrocatalytic OER, the surface oxidation of Co–P nanoparticles would likely result in more porous and hence a larger number of electrochemically active sites than what occurred in the case of Co/NC/CC. This observation is consistent with recent reports showing that metal phosphide-based electrocatalysts actually demonstrated better OER performance the corresponding metal oxides. The estimated ECSA values of Co–P/NC/CC, Co/NC/CC, ZIF-67/CC, and carbon cloth follow the same trend of their OER activity, emphasizing the critical role that ECSA plays in electrocatalysis. A detailed comparison of Co–P/NC/CC with other reported electrocatalysts for OER under strongly alkaline conditions can be found in Table S3,† suggesting the excellent performance of Co–P/NC/CC for OER applications.45–49 Noticeably, our Co–P/NC/CC could also rival the state-of-the-art OER catalyst IrO2 (Fig. S8b†), showing similar onset potential but much faster catalytic current rise.
 | ||
| Fig. 4 (a) LSV curves of Co–P/NC/CC (purple), Co/NC/CC (blue), ZIF-67/CC (red), and CC (black) for OER in 1.0 M KOH at a scan rate of 2 mV s−1. (b) Tafel plots of Co–P/NC/CC (purple), Co/NC/CC (blue), ZIF-67/CC (red), and CC (black) for OER. (c) EIS curves of Co–P/NC/CC (purple), Co/NC/CC (blue), ZIF-67/CC (red), and CC (black) measured at 1.573 V vs. RHE. Inset shows the expanded region around the intercepts at the x axis. (d) Scan rate dependence of the current densities of Co–P/NC/CC (purple), Co/NC/CC (blue), ZIF-67/CC (red), and CC (black) at 1.273 V vs. RHE. | ||
In order to demonstrate the fact that directly growing ZIF-67 on carbon cloth is crucial towards the excellent electrocatalytic performance of Co–P/NC/CC for both HER and OER, we conducted the following control experiments. ZIF-67 nano-polyhedrons were carbonized and phosphatized in a similar fashion as that of ZIF-67/CC. Subsequently, the resulting cobalt phosphide(s)-imbedded and nitrogen containing carbon matrices were loaded on a carbon cloth with Nafion as the polymer binder to produce a control sample Co–P/NC–CC. As shown in Fig. S9,† the HER and OER performances of Co–P/NC–CC are inferior to that of Co–P/NC/CC with same mass loading of cobalt. The catalytic current of Co–P/NC–CC did not take off until scanning to −0.3 V vs. RHE for HER and 1.6 V vs. RHE for OER, both of which required a larger overpotential compared to Co–P/NC/CC. In addition, the stability of our Co–P/NC/CC relative to CC for OER was also inspected. As shown in Fig. S10,† although CC was not stable for long-term OER electrolysis, our Co–P/NC/CC manifested its perfect stability in 1.0 M KOH for 50 h at 10 mA cm−2, showing unchanged chronopotentiometric curve over the entire course of OER electrolysis. These results unambiguously prove the effectiveness of anchoring ZIF-67 on carbon cloth during the preparation stage in producing more active and robust electrocatalysts.
With the excellent HER and OER performance of Co–P/NC/CC discussed above, we subsequently sought to fabricate a two-electrode electrolyzer for overall water splitting with same type catalyst-loaded carbon cloth electrodes serving as both cathode and anode in 1.0 M KOH. The LSV curves of these two-electrode systems were compared in Fig. 5a. The Co–P/NC/CC catalyst couple exhibited a catalytic onset potential less than 1.7 V. In fact, this catalyst couple approached current densities of 10 and 100 mV cm−2 at cell voltages of 1.77 and 1.93 V, respectively. In sharp contrast, the control samples of Co/NC/CC, Co–P/NC–CC, and ZIF-67/CC required a cell voltage of 1.89, 1.95, and 2.03 V, respectively, to arrive at 10 mA cm−2. As expected, carbon cloth was least active for water splitting. 20 h chronopotentiometry experiments were conducted to assess the stability of these catalyst couples for extended function at a catalytic current of 10 mA cm−2 (Fig. 5b). The Co–P/NC/CC catalyst couple required a voltage of ∼1.85 V and maintained this voltage throughout the entire course of electrolysis. All the other catalyst samples needed voltages at least 100 mV larger than that of Co–P/NC/CC. After the long-term chronoamperometry experiment, the composition and morphology of Co–P/NC/CC as cathode (Fig. S11†) and anode (Fig. S12†) investigated via SEM and elemental mapping analysis. Overall, the anchored cobalt phosphide(s)-based polyhedrons were still observed on the surface of carbon cloth with the anticipated distribution of Co, P, N, and C. A significant amount of oxygen was observed when Co–P/NC/CC acted as the anode, in agreement with the partial oxidation of cobalt phosphide(s)/cobalt during electrocatalytic OER.
 | ||
| Fig. 5 (a) LSV curves of Co–P/NC/CC (purple), Co/NC/CC (blue), Co–P/NC-CC (green), ZIF-67/CC (red), and CC (black) served as both cathode and anode in a two-electrode configuration at a scan rate of 2 mV s−1 in 1.0 M KOH for overall water splitting. (b) Chronopotentiometric curves of Co–P/NC/CC (purple), Co/NC/CC (blue), Co–P/NC–CC (green), ZIF-67/CC (red), and CC (black) in 1.0 M KOH at a current density of 10 mA cm−2 for 20 h. | ||
4. Conclusions
In summary, we described a facile route to synthesize self-supported and MOF-derived Co–P/NC/CC on carbon cloth as an effective electrocatalyst for overall water splitting. The as-prepared Co–P/NC/CC exhibited remarkable catalytic performance for both HER and OER under alkaline conditions with Tafel slopes of 52 and 61 mV dec−1, respectively. When employed as both anode and cathode for overall water splitting test, Co–P/NC/CC exhibited promising activity and robustness. Compared with other control samples, including ZIF-67/CC, Co/NC/CC, and Co–P/NC–CC, the excellent electrocatalytic activity of Co–P/NC/CC is mainly due to the competent active site (Co–P), highly porous and conductive substrate (NC and CC), and more importantly the intimate anchoring of Co–P/NC on carbon cloth. It is anticipated that such a convenient direct growing of MOF-derived catalysts on conductive substrate will open a new avenue for the development of promising electroactive materials for various applications.Acknowledgements
This work was supported by Utah State University (USU). X. L. acknowledges the financial support from the Chinese Academy of Sciences Funded Overseas Study Program. X. L. and J. D. thank the support of Qinghai Institute of Salt Lakes, Chinese Academy of Sciences. Y. S. also acknowledges the Microscopy Core Facility at USU for the SEM work.Notes and references
- D. Nocera, Chem. Soc. Rev., 2009, 38, 13 RSC.
- H. Gray, Nat. Chem., 2009, 1, 7 CrossRef CAS PubMed.
- M. Shalom, D. Ressnig, X. Yang, G. Clavel, T. Fellinger and M. Antonietti, J. Mater. Chem. A, 2015, 3, 8171 CAS.
- D. Voiry, H. Yamaguchi, J. Li, R. Silva, D. Alves, T. Fujita, M. Chen, T. Asefa, V. Shenoy, G. Eda and M. Chhowalla, Nat. Mater., 2013, 12, 850 CrossRef CAS PubMed.
- M. Gong, W. Zhou, M. Kenney, R. Kapusta, S. Cowley, Y. Wu, B. Lu, M. Lin, D. Wang, J. Yang, B. Hwang and H. Dai, Angew. Chem., Int. Ed., 2015, 127, 12157 CrossRef.
- Y. Jin, H. Wang, J. Li, X. Yue, Y. Han, P. Shen and Y. Cui, Adv. Mater., 2016, 28, 3785 CrossRef CAS PubMed.
- L. Trotochaud, S. Young, J. Ranney and S. Boettcher, J. Am. Chem. Soc., 2014, 136, 6744 CrossRef CAS PubMed.
- J. Chen, L. Dang, H. Liang, W. Bi, J. Gerken, S. Jin, E. Alp and S. Stahl, J. Am. Chem. Soc., 2015, 137, 15090 CrossRef CAS PubMed.
- O. Diaz-Morales, D. Ferrus-Suspedra and M. Koper, Chem. Sci., 2016, 7, 2639 RSC.
- B. You, N. Jiang, M. Sheng and Y. Sun, Chem. Commun., 2015, 51, 4252 RSC.
- N. Jiang, L. Bogoev, M. Popova, S. Gul, J. Yano and Y. Sun, J. Mater. Chem. A, 2014, 2, 19407 CAS.
- N. Jiang, Q. Tang, M. Sheng, B. You, D.-E. Jiang and Y. Sun, Catal. Sci. Technol., 2016, 6, 1077 CAS.
- B. You, N. Jiang and Y. Sun, Inorg. Chem. Front., 2016, 3, 279 RSC.
- B. You and Y. Sun, Adv. Energy Mater., 2016, 6, 1502333 CrossRef.
- A. Carim, F. Saadi, M. Soriaga and N. Lewis, J. Mater. Chem. A, 2014, 2, 13835 CAS.
- Y. Zheng, M. Gao, Z. Yu, Q. Gao, H. Gao and S. Yu, Chem. Sci., 2015, 6, 4594 RSC.
- B. You, N. Jiang, M. Sheng, M. Bhushan and Y. Sun, ACS Catal., 2015, 5, 714 Search PubMed.
- J. Ryu, N. Jung, J. Jang, H. Kim and S. Yoo, ACS Catal., 2015, 5, 4066 CrossRef CAS.
- N. Jiang, B. You, M. Sheng and Y. Sun, ChemCatChem, 2016, 8, 106–112 CrossRef CAS.
- K. Xu, P. Chen, X. Li, Y. Tong, H. Ding, X. Wu, W. Chu, Z. Peng, C. Wu and Y. Xie, J. Am. Chem. Soc., 2015, 137, 4119 CrossRef CAS PubMed.
- T. Ma, S. Dai, M. Jaroniec and S. Qiao, Angew. Chem., Int. Ed., 2014, 53, 7281 CrossRef CAS PubMed.
- W. Zhou and L. Guo, Chem. Soc. Rev., 2015, 44, 6697 RSC.
- Y. Liu, H. Cheng, M. Lyu, S. Fan, Q. Liu, W. Zhang, Y. Zhi, C. Wang, C. Xiao, S. Wei, B. Ye and Y. Xie, J. Am. Chem. Soc., 2014, 136, 15670 CrossRef CAS PubMed.
- E. Hernández-Pagán, N. Vargas-Barbosa, T. Wang, Y. Zhao, E. Smotkin and T. Mallouk, Energy Environ. Sci., 2012, 5, 7582 Search PubMed.
- C. Tang, N. Cheng, Z. Pu, W. Xing and X. Sun, Angew. Chem., Int. Ed., 2015, 54, 9351 CrossRef CAS PubMed.
- H. Wang, H. Lee, Y. Deng, Z. Lu, P. Hsu, Y. Liu, D. Lin and Y. Cui, Nat. Commun., 2015, 6, 7261 CrossRef CAS PubMed.
- B. You, N. Jiang, M. Sheng, S. Gul, J. Yano and Y. Sun, Chem. Mater., 2015, 27, 7636 CrossRef CAS.
- B. You, N. Jiang, M. Sheng, W. Drisdell, J. Yano and Y. Sun, ACS Catal., 2015, 5, 7068 CrossRef CAS.
- Y. Luo, J. Jiang, W. Zhou, H. Yang, J. Luo, X. Qi, H. Zhang, D. Yu, C. Li and T. Yu, J. Mater. Chem., 2012, 22, 8634 RSC.
- J. Roy-Mayhew, G. Boschloo, A. Hagfeldt and I. Aksay, ACS Appl. Mater. Interfaces, 2012, 4, 2794 CAS.
- N. Cheng, Q. Liu, J. Tian, Y. Xue, A. Asiri, H. Jiang, Y. He and X. Sun, Chem. Commun., 2015, 51, 1616 RSC.
- Z. Zhao, H. Wu, H. He, X. Xu and Y. Jin, Adv. Funct. Mater., 2014, 24, 4698 CrossRef CAS.
- J. Tian, Q. Liu, A. Asiri and X. Sun, J. Am. Chem. Soc., 2014, 136, 7587 CrossRef CAS PubMed.
- J. Wang, H. Zhong, Z. Wang, F. Meng and X. Zhang, ACS Nano, 2016, 10, 2342 CrossRef CAS PubMed.
- P. Wang, F. Song, R. Amal, Y. Ng and X. Hu, ChemSusChem, 2016, 9, 472 CrossRef CAS PubMed.
- W. Fang, D. Liu, Q. Lu, X. Sun and A. Asiri, Electrochem. Commun., 2016, 63, 60 CrossRef CAS.
- W. Zhang, Z. Wu, H. Jiang and S. Yu, J. Am. Chem. Soc., 2014, 136, 14385 CrossRef CAS PubMed.
- J. Yang, F. Zhang, H. Lu, X. Hong, H. Jiang, Y. Wu and Y. Li, Angew. Chem., Int. Ed., 2015, 54, 10889 CrossRef CAS PubMed.
- H. Li, P. Yang, D. Chu and H. Li, Appl. Catal., A, 2007, 325, 34 CrossRef CAS.
- A. Grosvenor, S. Wik, R. Cavell and A. Mar, Inorg. Chem., 2005, 44, 8988 CrossRef CAS PubMed.
- H. Zhang, Z. Ma, J. Duan, H. Liu, G. Liu, T. Wang, K. Chang, M. Li, L. Shi, X. Meng, K. Wu and J. Ye, ACS Nano, 2016, 10, 684 CrossRef CAS PubMed.
- H. Jin, J. Wang, D. Su, Z. Wei, Z. Pang and Y. Wang, J. Am. Chem. Soc., 2015, 137, 2688 CrossRef CAS PubMed.
- H. Vrubel and X. Hu, Angew. Chem., Int. Ed., 2012, 51, 12703 CrossRef CAS PubMed.
- L. Feng, H. Vrubel, M. Bensimon and X. Hu, Phys. Chem. Chem. Phys., 2014, 16, 5917 RSC.
- C. McCrory, S. Jung, I. Ferrer, S. Chatman, J. Peters and T. Jaramillo, J. Am. Chem. Soc., 2015, 137, 4347 CrossRef CAS PubMed.
- N. Jiang, B. You, M. Sheng and Y. Sun, Angew. Chem., Int. Ed., 2015, 54, 6251 CrossRef CAS PubMed.
- Y. Li, P. Hasin and Y. Wu, Adv. Mater., 2010, 22, 1926 CrossRef CAS PubMed.
- J. Ren, M. Antonietti and T. Fellinger, Adv. Energy Mater., 2015, 5, 1401660 CrossRef.
- F. Song and X. Hu, Nat. Commun., 2014, 5, 4477 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional electrochemistry and characterization data, Fig. S1–S12 and Tables S1–S3. See DOI: 10.1039/c6ra17030g |
| This journal is © The Royal Society of Chemistry 2016 |