
Synthesis, optical, and chemical properties of a π-extended rhodol derivative and its derivatives with selectivity and sensitivity for sensing Hg2+ in aqueous media†
Abstract
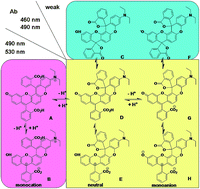
A π-extended rhodol derivative 1 with five fused six-membered rings has been synthesized. As the optical properties of the dye 1 are strictly controlled by two spirorings within the structure, its reversible structural conversions among the monocation, neutral, and monoanion forms were investigated in detail under different conditions by the electronic absorption and fluorescence emission spectroscopy. Then the chemical modification methodology of dye 1 was investigated in detail. The phenol hydroxyl could be replaced by a chlorine atom by using POCl3 as a chlorination reagent. Only one of the carboxyl groups could be esterificated by alcohol under strong acidic conditions. Finally, a thiosemicarbazide spirolactam 7 was obtained from dye 1 as a highly Hg2+-selective fluorescent chemodosimeter.
 Please wait while we load your content...
Please wait while we load your content...

