
Temperature-dependent photoluminescence of inorganic perovskite nanocrystal films
Abstract
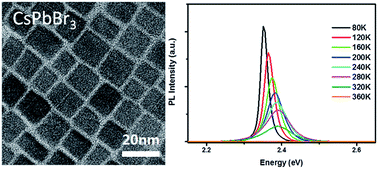
Temperature-dependent photoluminescence (PL) properties of inorganic perovskite CsPbBr3 nanocrystal (NC) films were studied by using steady-state and time-resolved PL spectroscopy. The closely packed solid films were obtained by dropping NC solution on silicon substrates. It was found that the PL intensities of the NC films, which are dependent on the size of NCs, slightly decreased with increasing temperature to 300 K, while the PL intensities dropped rapidly with increasing temperature above 300 K and were nearly quenched at 360 K. Further the corresponding average PL lifetimes increased significantly with increasing temperature below about 320 K and then significantly became shorter. The PL quenching mechanisms were demonstrated through heating and cooling experiments. The experimental results indicated inorganic perovskite NCs exhibited a thermal PL quenching in the temperature range of 80–300 K and a thermal degradation at temperatures above 300 K. The linewidths, peak energies, and lifetimes of PL emissions for the NC films as a function of temperature were discussed in detail.
 Please wait while we load your content...
Please wait while we load your content...

