
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Vapor-based micro/nano-partitioning of fluoro-functional group immobilization for long-term stable cell patterning†
Shun-ichi Funano,
Nobuyuki Tanaka and
Yo Tanaka*
Quantitative Biology Center (QBiC), RIKEN, 1-3 Yamadaoka, Suita, Osaka 565-0871, Japan. E-mail: yo.tanaka@riken.jp
First published on 11th October 2016
Abstract
A technique for partitioning cell adhesion with long-term stability is an important tool in biology and medical experiments. This study has developed a simple vapor-based immobilization method using a compound bearing a fluoro-functional group on a cell culture surface with micro/nanoscale patterns. The vapor of a silane coupling agent containing the fluoro-functional group was introduced into the micro-/nanochannels of polydimethylsiloxane elastomer placed on the target surface in a vacuum desiccator. The agent was immobilized with a pattern shown only on the areas of the channels on the surface. Although it is difficult to introduce the agent solution into a sub-micrometer-width channel via a conventional liquid-based approach, the vapor can be introduced into nanochannels with an exceptionally narrow width of 800 nm and an exceptionally low height of 700 nm. The fluoro-functional group area was clearly found to repel the adhesion of fluorescent-dye-conjugated fibronectin molecules. Four different types of cells, mouse skeletal myoblast cell line C2C12 cells, human cervix adenocarcinoma cell line HeLa cells, bovine artery endothelial cells HH, and mouse mammary gland epithelial cell line NMuMG cells, were prepared and each type was seeded onto the immobilized culture surfaces of glass and polystyrene dishes. The cells were adhered on the non-fluoro-functional group immobilized area and the cell pattern was maintained during atleast a 39 days of cultivation. This method is expected to be useful for simple and stable cell patterning in biological and medical fields.
Introduction
Behavior is regulated in a controlled cell adhesion, especially with respect to cell morphology and function. Different areas of a cell adhesion region offer dramatic changes in cell fate; for example, cell death on a small area and high cell activity on a larger area.1 Cells are well-known to be highly sensitive to adherent patterns.2–4 To understand the cell behavior in a regulated cell adhesion, methods for cell patterning have been used. In particular, micro-contact printing5–9 is a simple and popular method to transfer molecules for cell adhesion from a polydimethylsiloxane (PDMS) stamp surface to a target surface via contact between the PDMS stamp and the target. The resolution of patterns for micro-contact printing has become on a sub-micron level. Similarly, striped PDMS microchannels10 or laminar flow11 have also been used for patterning molecules, but these protein based cell patterning methods have a lack of long-term stability because they are based on physical adsorption onto a target surface.When compared to these methods, hydrogel-based patterning methods have long-term stability. These involve negative patterning, which is based on the spatial distribution of repellency to cells using non-cell adhesive polymers.12–16 The advantage of these hydrogel-based patterning methods is the long-term stability provided, generally more than two weeks. However, the methods require dedicated apparatus such as an electron beam exposure device,17 an ink-jet device,18 laser,19,20 a UV light irradiation source12 or complicated chemical operations12 every time the patterning experiments are carried out. Hydrogel patterning methods using a PDMS stencil,21,22 milling and three-dimensional printer are more simple. However, due to their fabrication limitations, the resolution is typically more than 50 μm. Therefore, it has been difficult to control the shape of cells, which is required in practical biological studies. The authors have also reported an agarose gel casting method for degassed PDMS molds.23 In this casting method, an agarose solution is aspirated into micro-channels, dehydrated and cast to make gel patterns. However, the gel structure is actually still larger than about 20 μm because the viscosity of the agarose solution reduces the fluidity of the solution in the micro-channels. In addition, the long-term stability of this physical adsorption-based gel patterning has been reported to be typically around 10 days.21,24 This characteristic sometimes causes observation problems, for both long-term observations (more than 10 days) and for high-resolution observations of a single cell.25
To address these issues, this study developed a gas-phase micro/nanopatterning method using the vapor of a silane coupling agent containing a fluoro-functional group instead of hydrogel using PDMS micro-channels for partitioning silanization (Fig. 1). Because of its low viscosity, the vapor can be introduced into a narrow space, even a nano-sized channel. Moreover, chemical-bond-based immobilization using the silane coupling agent guarantees the long-term stability. Based on this background, the aim of this study was to demonstrate vapor-based surface modification for cell patterning. First, the principle and resolution of patterned immobilization of the compound bearing a fluoro-functional group were verified. Then, the immobilized surfaces were applied to cell culturing and the long-term stability of the cell pattern was demonstrated.
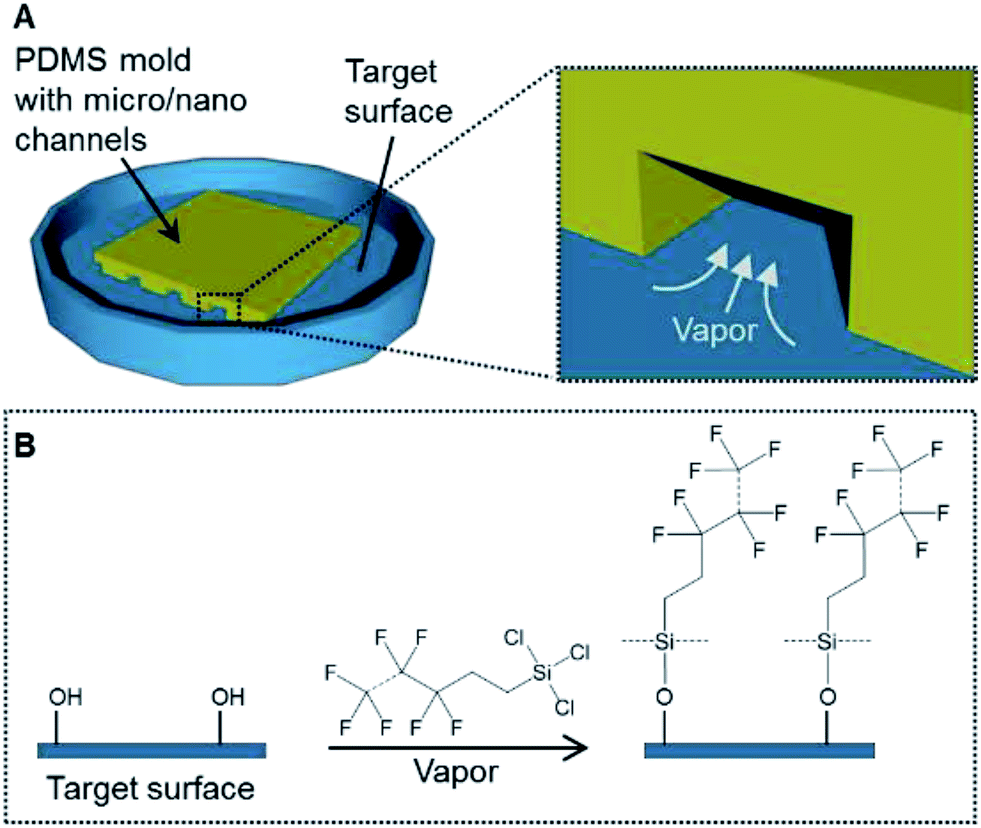 | ||
| Fig. 1 (A) The concept of a patterning method using the vapor of a silane coupling agent. (B) A schematic of the silanization process on the target surface. | ||
Experimental
Preparation of the PDMS molds
A polydimethylsiloxane (PDMS) (SILPOT 184) (Dow Corning Toray, Tokyo, Japan) mold with micro-grooves was prepared following a reported procedure.26 Briefly, a glass plate with a stripe structure was prepared using a dicing saw to make grooves with a depth of 300 μm, a width of 300 μm and a pitch of 500 μm. For a PDMS mold with nano-grooves, a silicon wafer with a striped structure was prepared using electron-beam lithography to make grooves with a depth of 700 nm, a width of 800 nm and a pitch of 1700 nm.27 The PDMS pre-polymer and a curing agent were mixed at a ratio of 10 to 1. The PDMS mixture was poured into each mold and cured at 70 °C for 3 h. The cured PDMS elastomer was peeled off the glass plate or the silicon wafer. This cured PDMS elastomer was further hardened at 150 °C for 3 h. Then, the PDMS mixture was poured into the hardened PDMS elastomer and cured at 70 °C for 3 h. This cured PDMS elastomer was peeled off the hardened PDMS elastomer. The obtained PDMS elastomer was used as the PDMS mold with micro or nano-grooves for the following experimental steps (Fig. 2A and B). The surface profiles of the PDMS molds were measured with a three-dimensional laser scanning confocal microscope (VK-8710, Keyence, Osaka, Japan).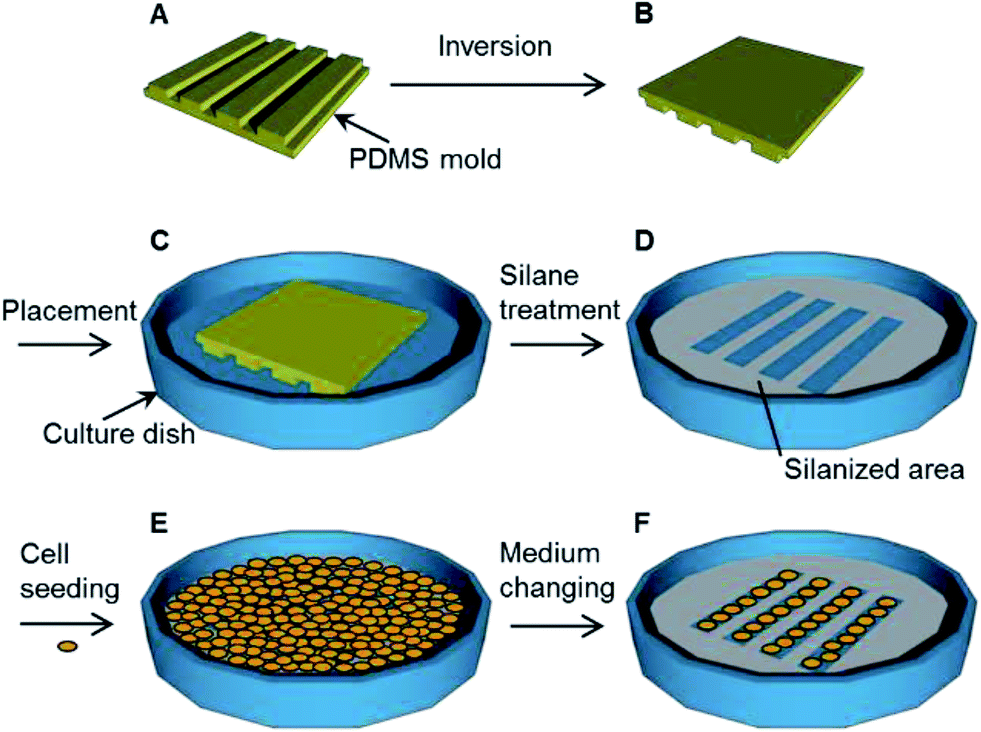 | ||
| Fig. 2 The procedure used for the immobilization of a pattern using the vapor of a silane coupling agent. (A) Fabrication of the polydimethylsiloxane (PDMS) mold. (B) Inversion of the PDMS mold. (C) Placement of the PDMS mold on the target surface. (D) Treatment with the silane coupling agent vapor and removal of the PDMS mold. (E) Seeding of cells. (F) Changing of the culture medium after a brief incubation, followed by further cell cultivation. | ||
Patterned immobilization
Glass bottom dishes (D11140H) (Matsunami Glass Industry, Osaka, Japan) and polystyrene dishes (353001) (Thermo Fisher Scientific, Waltham, MA, USA) were treated with vacuum oxygen plasma at an intensity of 30 W and oxygen flow rate of 8 mL min−1 for 2 min in the chamber of a compact etcher (FA-1, SAMCO, Kyoto, Japan). The PDMS molds were immersed into deionized water for 5 s and dried with flowing nitrogen gas. Each PDMS mold was placed on a culture dish. The spaces between the dish surface and the grooves of the PDMS mold formed the micro- or nano-channels. Each PDMS mold was pressed with a 1 N weight for 2 min (Fig. 2C). The dishes with the PDMS molds were placed in a vacuum desiccator (volume, 90 mL) and then exposed to the vapor produced from 50 μL of trichloro(1H,1H,2H,2H-heptadecafluorodecyl)silane (T2705) (Tokyo Chemical Industry Tokyo, Japan) while evacuating the desiccator for 1 h in order to immobilize the fluoro-functional group (Fig. 2D).7 After the PDMS molds were removed from the dishes, these dishes were sterilized under UV light at 254 nm for 1 h. The contact angles of the outside area where the PDMS mold was placed were measured with a contact angle meter (DMs-401, Kyowa Interface Science, Saitama, Japan).Fibronectin application
Non-fluorescent fibronectin (F1141) (Sigma-Aldrich, St. Louis, MO, USA) was applied onto the PDMS mold surface following a reported procedure.7,28 Briefly, the PDMS molds were sequentially washed with acetone, 2-propanol, ethanol and deionized water, and then dried with flowing nitrogen gas. The PDMS molds were treated with vacuum oxygen plasma at intensity of 30 W and oxygen flow rate of 8 mL min−1 for 2 min in the chamber of the compact etcher. The PDMS molds were placed in the vacuum desiccator and then exposed to the vapor produced by 10 μL of trichloro(1H,1H,2H,2H-heptadecafluorodecyl)silane while evacuating the desiccator for 1 h. After exposure to the silane vapor, 10 μg mL−1 non-fluorescent fibronectin Dulbecco's phosphate buffer saline (PBS) (11482-15) (NACALAI TESQUE, Kyoto, Japan) solution was applied to the PDMS molds for 1 h. The PDMS molds were dried at room temperature.Assessment for the adsorption and transfer of fibronectin
In the case of the adsorption of fibronectin, to a culture dish was placed a PDMS mold with nano-grooves (Fig. 3A and B) and the dish was silanized (Fig. 3C). After the PDMS mold was removed from the dish, 1 mg mL−1 rhodamine fibronectin (FNR01) (Cytoskeleton, Denver, CO, USA) PBS solution was applied to the dish for 1 h at the area where the PDMS mold was placed (Fig. 3D). The dish was washed three times with PBS solution and then dried with flowing nitrogen gas (Fig. 3E). In the case of the transfer of fibronectin, a 1 mg mL−1 rhodamine fibronectin PBS solution was applied to a PDMS mold with micro-grooves for 1 h and this was allowed to dry at room temperature (Fig. 3F and G). The PDMS mold was placed on a culture dish (Fig. 3H). After the dish was silanized, the PDMS mold was removed from the dish (Fig. 3I). Fluorescence images were observed with an inverted system microscope (IX71, Olympus, Tokyo, Japan) equipped with a charge-coupled device (CCD) camera (24-bit RGB color) and filters (excitation filter: 530–550 nm (bandpass); emission filter: 575 nm (longpass)).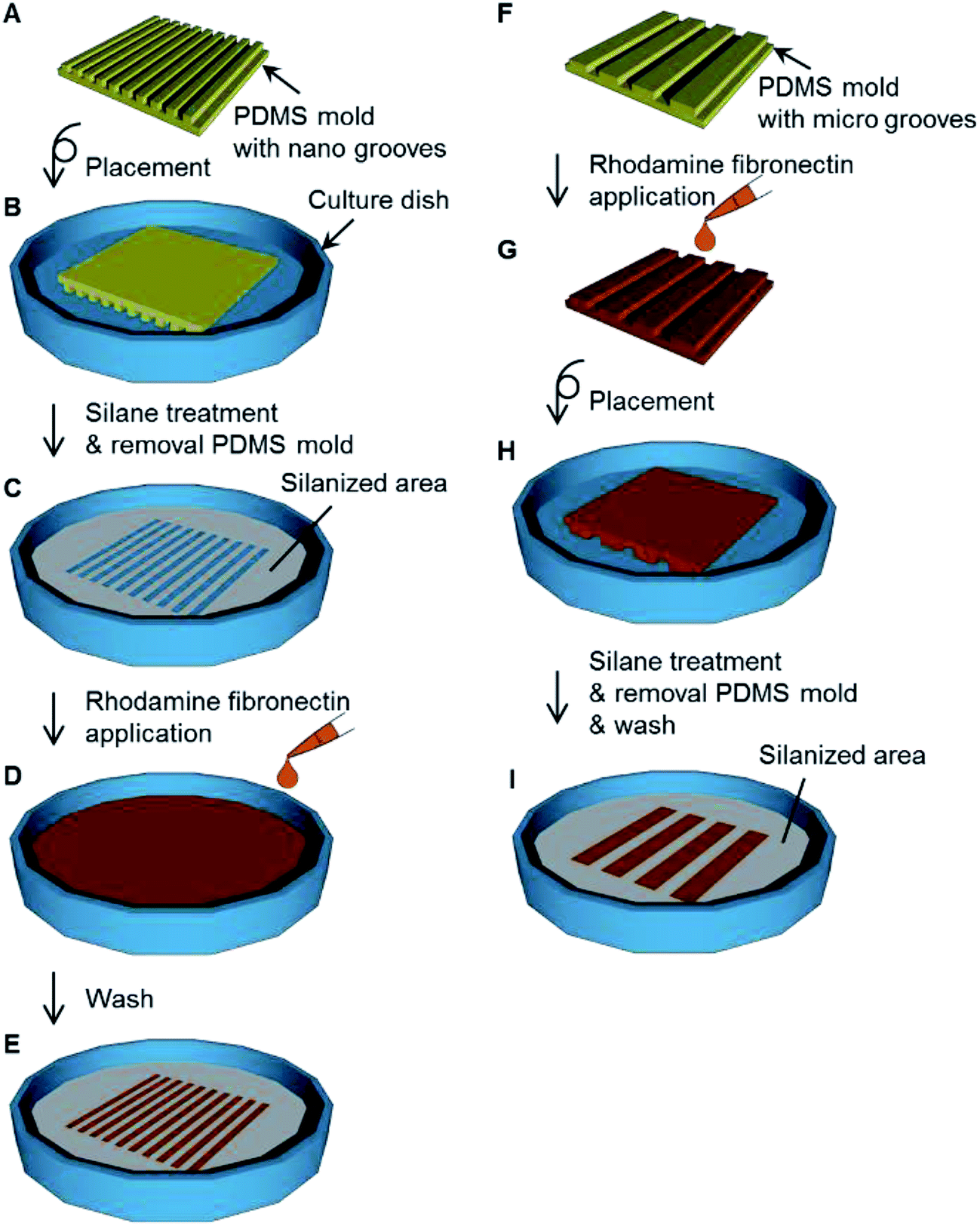 | ||
| Fig. 3 Procedure for the assessment for the adsorption and transfer of fibronectin. (A) Fabrication of polydimethylsiloxane (PDMS) mold with nano-grooves. (B) Placement of the PDMS mold on the culture dish. (C) Treatment with the silane coupling agent vapor. (D) Adsorption of rhodamine fibronectin onto the culture dish. (E) Patterned adsorption of fibronectin. (F) Fabrication of PDMS mold with micro-grooves. (G) Application of rhodamine fibronectin onto the PDMS mold. (H) Placement of the PDMS mold on the culture dish. (I) The dish with the pattern of silanized area and fibronectin transferred from the PDMS mold. | ||
Cell culture
Mouse skeletal myoblasts cell line C2C12 cells, human cervix adenocarcinoma cell line HeLa cells, bovine artery endothelial cells HH, and mouse mammary gland epithelial cell line NMuMG cells were cultivated in Dulbecco's modified Eagle's medium (D6429) (Sigma-Aldrich, St. Louis, MO, USA) with 10 v/v% calf serum (12133C) (Nichirei Biosciences, Tokyo, Japan) and 1 v/v% penicillin and streptomycin antibiotics (168-23191) (Wako Pure Chemical Industries, Osaka, Japan) in a dish (Fig. 2E). The cultivation was conducted at 37 °C under humidified conditions with 5% CO2 for 3 h. After 3 h of cultivation, the supernatant was changed with fresh culture medium to remove the floating cells (Fig. 2F). The supernatant was changed with fresh culture medium every 3 d during the subsequent cultivation. Cells were observed with the inverted system microscope. The observed images were analyzed using ImageJ software.Results and discussion
Immobilization of compound with a fluoro-functional group by vapor phase silanization
The contact angle of the glass bottom dishes and polystyrene dishes changed from approximately 75° to 110° after vapor phase silanization for 1 h (Fig. 4). This result suggested that the dish surface characteristics changed from hydrophilic to hydrophobic. This was attributed to the effect of the fluoro-functional group derived from the silane coupling agent, which was immobilized onto the dish surface because the contact angles after the vapor silanization almost coincided with the contact angle of a fluoro-functionalized surface previously reported.29 In addition, the whole bottom surface (56 cm2) was treated via a single-step procedure because the silane coupling agent vapor filled the surrounding space of the target dishes. Therefore, this vapor phase silanization method was considered to be applicable to the treatment of large area surface or large numbers of target surfaces. Moreover, when compared to typical chemical bonding of non-cell adhesive polymer requiring 5 steps in every cell culture experiments,14 the simplicity has dramatically increased.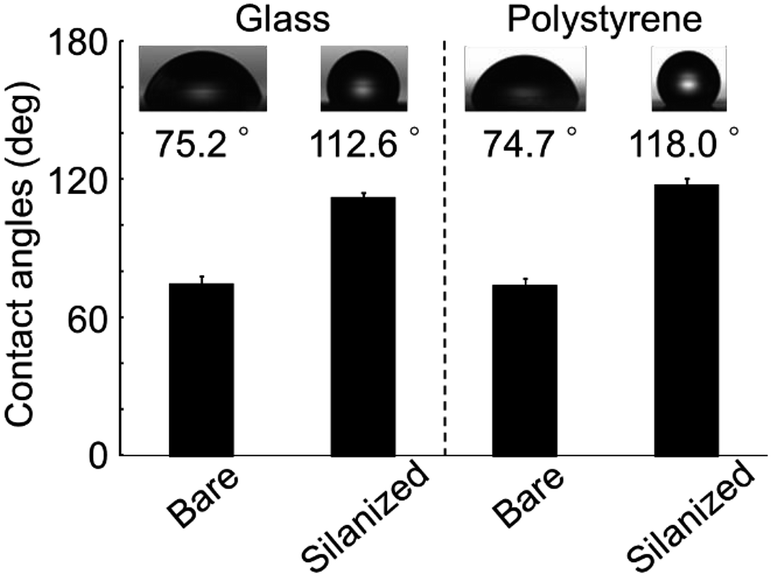 | ||
| Fig. 4 The contact angles on the bare or silanized cell culture surfaces of both glass and polystyrene. The photographs in the graph show droplets of deionized water on the surfaces. The data bars and error bars indicate the mean values and standard deviations, respectively (n = 5). | ||
Patterned immobilization
A PDMS mold with nano-grooves was successfully obtained (Fig. 5A and B). To assess the adsorption onto the patterned immobilized surface, the fluorescence intensity was monitored (Fig. 5C). The stripe of adsorbed rhodamine fibronectin corresponding to the profile of the PDMS mold was clearly observed. This result suggested that the fluoro functional group was immobilized with the striped pattern on the surface by permeation of vapor via the nano-channels. Because the vapor viscosity is generally lower than that of a liquid, especially an agarose solution,23 the permeability of vapor into the nano-spaces was increased in comparison to the liquid, resulting in nano-patterned immobilization. This result demonstrated that this method was feasible for patterned immobilization with nano-scale resolution. This method can improve the patterning resolution when compared with the previously reported resolution of approximately 20 μm.23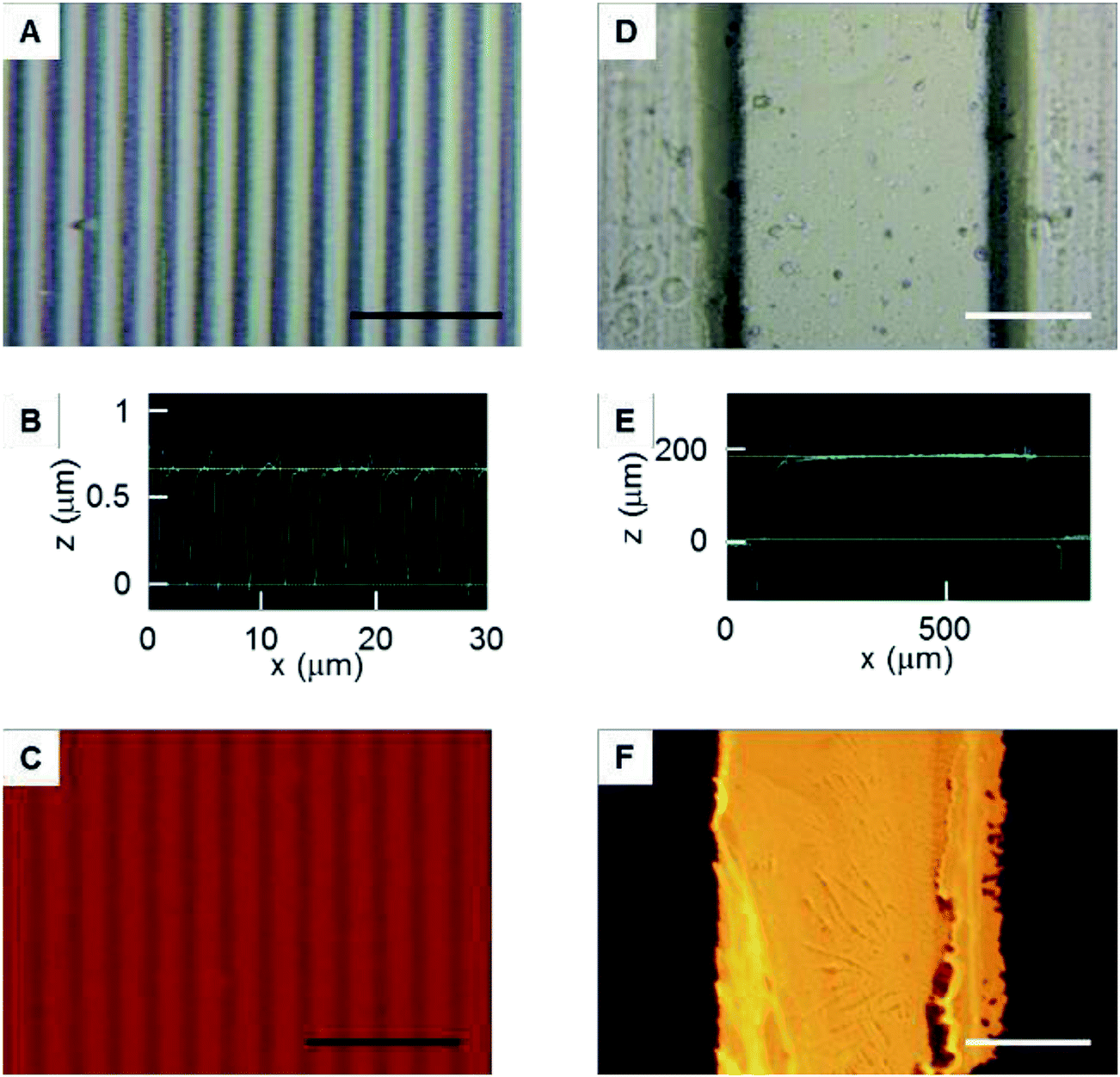 | ||
| Fig. 5 Patterned immobilization. (A) Microphotograph of the polydimethylsiloxane (PDMS) mold with nano-grooves. (B) The cross-sectional shapes of the PDMS mold with nano-grooves. (C) The adsorption of fluorescent fibronectin onto the patterned immobilized surface. (D) Microphotograph of the PDMS mold with micro-grooves. (E) The cross-sectional shapes of the PDMS mold with micro-grooves. (F) Transfer of fluorescent fibronectin from the PDMS mold to the cell culture surface. The black and white bars indicate 10 μm and 200 μm lengths, respectively. | ||
A PDMS mold with micro-grooves was also successfully obtained (Fig. 5D and E). To assess the transfer of fibronectin from the PDMS mold to the surfaces, the fluorescence intensity was monitored (Fig. 5F). The line of transferred rhodamine fibronectin corresponding to the profile of the PDMS mold was clearly observed. This result suggested that the method can be combined with micro-contact printing to transfer other functional molecules with a micro-pattern. The surface can be simultaneously immobilized with a pattern combining both cell-adhesive and cell-repellent molecules using the fibronectin transfer.
Efficacy for cell patterning
Each surface condition of the culture dish is illustrated (Fig. 6A). The morphologies of C2C12 cells cultivated on the dishes for 1 d were monitored (Fig. 6B–E). No cell pattern was observed on the dish without non-fluorescent fibronectin transfer or silanization (Fig. 6B). A poor cell pattern was observed on the dish without non-fluorescent fibronectin transfer but with silanization (Fig. 6C). No cell pattern was observed on the dish with non-fluorescent fibronectin transfer but without silanization (Fig. 6D). A clear pattern was observed on the dish with both non-fluorescent fibronectin transfer and silanization (Fig. 6E). Summarizing the above results, the generation of cell patterns was observed only in the case of silanization. On the other hand, cells were non-specifically adhered on the whole bottom surface of the dishes in the case of no silanization.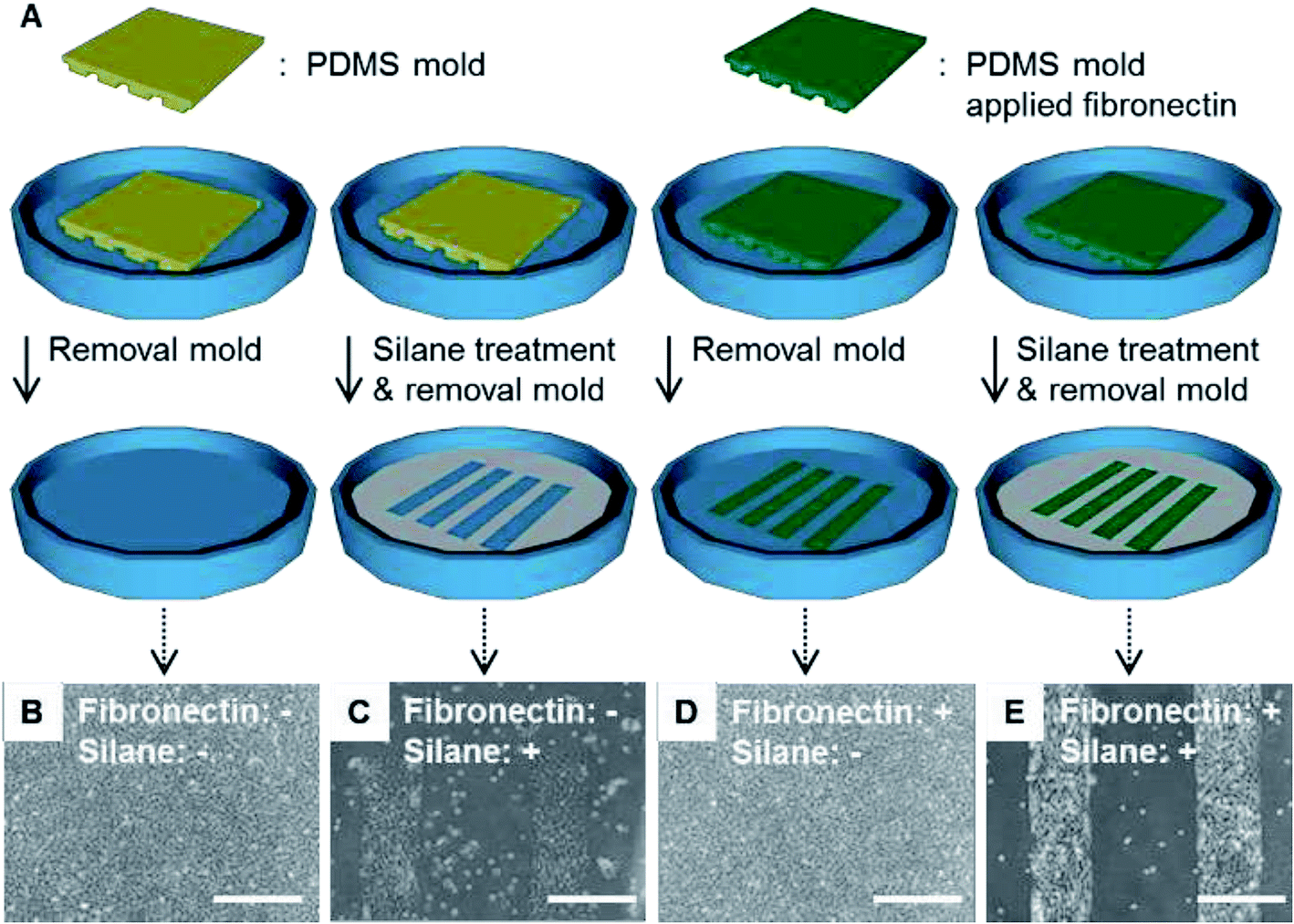 | ||
| Fig. 6 (A) Illustrations to show the surface conditions of the culture dishes. (B)–(E) Microphotographs showing the cells attached on the surfaces in the four cases. (B) Without non-fluorescent fibronectin or silane coupling agent. (C) Without non-fluorescent fibronectin, with silane coupling agent. (D) With non-fluorescent fibronectin, without silane coupling agent. (E) With non-fluorescent fibronectin and the silane coupling agent. The white bar indicates 500 μm length. | ||
These results suggest that the silanization was dominant for obtaining cell patterns when compared with the transfer of the extracellular matrix such as fibronectin. The silanization was dominant for obtaining cell patterns because the cells were adhered on the bare surface of the dish bottom. This experiment demonstrated that the patterned immobilization obtained by the vapor of a silane coupling agent was important for obtaining cell patterns.
Time dependency of silanization
The morphologies of the C2C12 cells cultured on the dishes silanized for 0, 1, 3 and 6 h were monitored (Fig. 7A–D). The silanization time of 0 h means without silanization. Cells adhered with a stripe pattern on the bottom of the dishes for silanization times of 1, 3 and 6 h. On the other hand, the cells adhered on the whole bottom surface of the dish for the silanization time of 0 h (without silanization).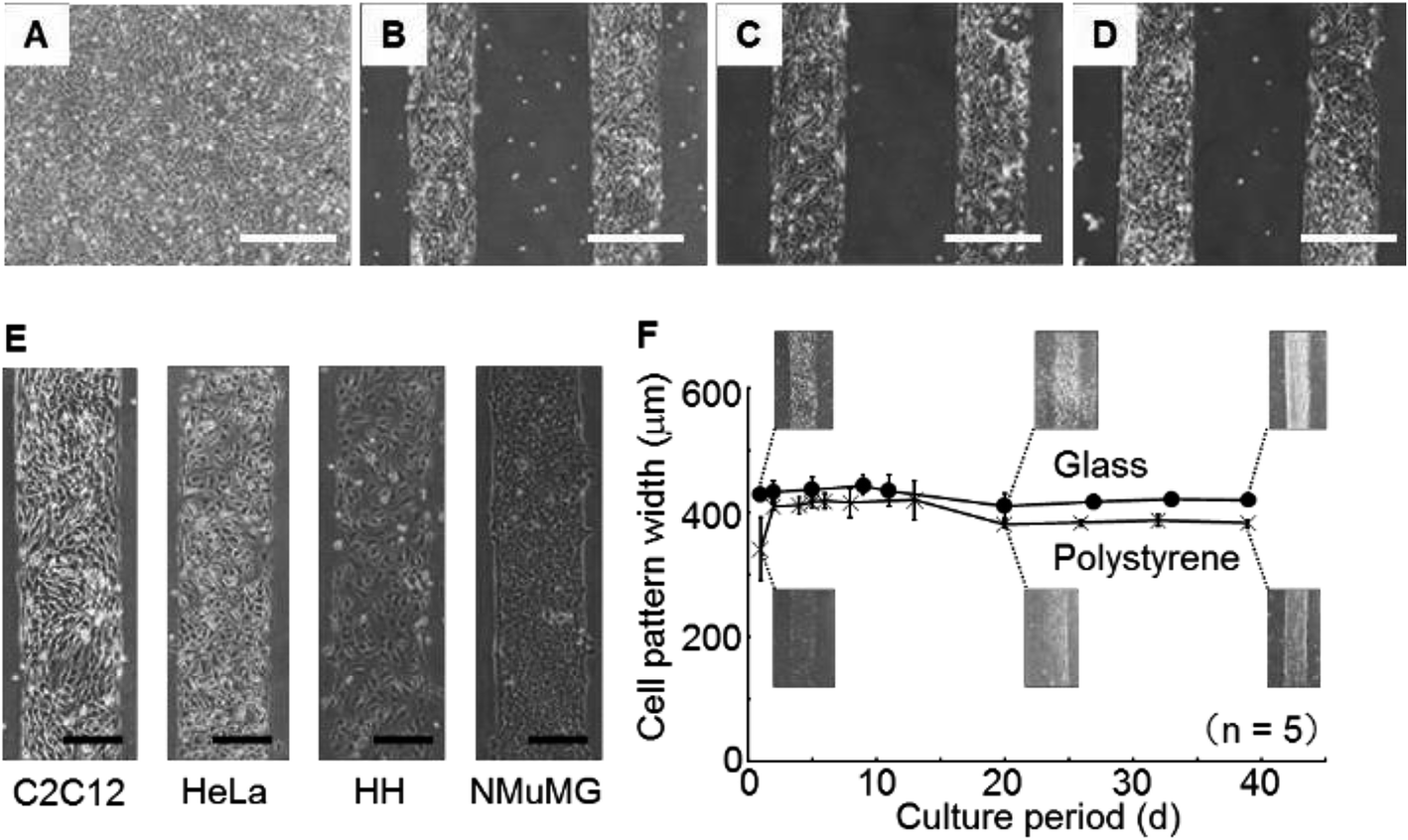 | ||
| Fig. 7 (A)–(D) Microphotographs showing the cells attached on the surfaces treated with the silane coupling agent for 0 h, 1 h, 3 h and 6 h, respectively. (E) Microphotographs of the cell patterns of the four types of cells. (F) Time courses of the cell pattern width: (●) glass bottom dish and (×) polystyrene dish. The white and black bars indicate 500 μm and 200 μm lengths, respectively. | ||
These results suggest that the silanization proceeded sufficiently for the reaction time of 1 h. Even if silanization was carried out for more than 3 h, more silane coupling agent was not able to bind covalently onto the dish surface because the effectiveness of the oxygen plasma processing, which was necessary for silanization, was dramatically lost within 2 h.30
Application to various cells
The morphologies of C2C12 cells, HeLa cells, HH and NMuMG cells cultivated on the dishes for 1 d were monitored (Fig. 7E). Clear patterns were observed independently of the cell type. These results suggested that the method would be sufficiently versatile for adhesive cells because the fluoro-functional group was a strong repellent to protein adsorption.Investigation of long-term stability
During 39 d of cultivation of C2C12 cells, the widths of the cell patterns on the glass bottom dishes or the polystyrene dishes were measured (Fig. 7F). The line widths of the cell patterns increased for 2 d in the polystyrene dishes. The decreasing widths of the line pattern composed of the cells was observed, beginning from 10 d and continuing to 20 d on the glass bottom and polystyrene dishes, thereafter, the pattern width was constant. After further cultivation, the patterns were maintained until 39 d.The initial increase in the line width on the polystyrene dishes during the initial 2 d of cultivation was thought to be caused by cellular proliferation reaching the boundary of the immobilized area of the silane coupling agent. This result suggested that the cells were unable to invade into the fluoro-functional group immobilized area. On the other hand, the width decreased from 10 d to 20 d because the cells fused and the cell clusters shrank. The cells were mouse skeletal myoblasts and they differentiate to myotubes, which are multi-nucleate cells.31 The decrease in cell pattern width seemed to be caused by increasing the extent of cell fusion in the differentiation from myoblasts to myotubes. Maintenance of the cell pattern result during more than 1 month of cultivation suggested that the stability was superior to that given by conventional micro-contact printing and other methods. In addition, the cytotoxicity of the fluoro-functional group was restrictive because the cell pattern was maintained during more than 1 month of cultivation.
Performance evaluation
Here, the performance of the presented method was compared with the previously reported methods from a viewpoint of long-term stability and resolution. As for the long-term stability, the developed method provided stable patterning for a maximum of 39 d, which was far longer than that observed for protein-based patterning (the stability was lost within a few days).21 Furthermore, because this method was based on chemical bonding instead of physical adsorption, long-term maintenance of the cell pattern was possible. There have been several methods based on chemical bonding for cell patterning, however, they have required micro-patterned photo-irradiation just before actual use for cell culturing. In the developed method, once the PDMS molds with desired patterns were obtained, patterned immobilization can be performed repeatedly without such special equipment as micro-patterned photo-irradiation devices. The silanization-based patterning method achieved a resolution of 800 nm, which was almost the same as the width of neuron dendrites. This was far smaller than that observed for the previously reported chemical-binding-based patterning using light irradiation (typically the resolution is over 10 μm).12,14–16This property can be advantageous for various bioanalytical fields; for example, it makes the observation of certain cellular phenomena possible in a relatively long time span, such as neural network formation of neurons32 or muscle cells33 and nucleus elongation34 at a single-cell level. Moreover, the simplicity of the developed method promotes such studies in biological laboratories. The silanization-based patterning can also be applied for single biomolecule analysis. For analysis of ultra-small sample volume samples such as single cells, chemical surface patterning in micro-/nanochannels or chambers has been used,35–39 but their patterning resolution was more than 10 μm. By applying the developed method, the resolution can be dramatically improved and therefore, large-integrated single-molecule level patterning and analysis becomes possible. Overall, the method is a strong potential tool for single-cell and single-molecule analyses in vitro.
Conclusions
The simple, easy and useful partitioning method was developed to immobilize a silane coupling agent. The fluoro-functional group was immobilized on the surface of dishes by permeation of vapor via the micro-/nanochannels. This method was combined with micro-contact printing to transfer other functional molecules. The silanization was the key point of this method and took 1 h. The method was found to be applicable to several types of adhesive cells. Large surface areas or large numbers of target surfaces were treated by a single-step procedure. The cell patterns were maintained during longer than 1 month of cultivation. This method is a simple, easy and inexpensive tool to achieve patterning with both high-resolution and stability without special equipment in life science laboratories.Conflict of interest
The authors have no conflict of interest directly relevant to the content of this article.Acknowledgements
This work was supported by Grants-in-Aid for Young Scientists (A) (25709081) and Young Scientists (B) (15K18004) from Japan Society for the Promotion of Science and the Funding Program for Impulsing PAradigm Change through disruptive Technologies (ImPACT) from the Cabinet Office, Government of Japan.References
- C. S. Chen, M. Mrksich, S. Huang, G. M. Whitesides and D. E. Ingber, Science, 1997, 276, 1425–1428 CrossRef CAS PubMed.
- S. N. Bhatia, U. J. Balis, M. L. Yarmush and M. Toner, FASEB J., 1999, 13, 1883–1900 CAS.
- S. A. Ruiz and C. S. Chen, Stem Cells, 2008, 26, 2921–2927 CrossRef PubMed.
- T. Takebe, K. Sekine, M. Enomura, H. Koike, M. Kimura, T. Ogaeri, R. Zhang, Y. Ueno, Y. Zheng, N. Koike, S. Aoyama, Y. Adachi and H. Taniguchi, Nature, 2013, 499, 481–484 CrossRef CAS PubMed.
- R. S. Kane, S. Takayama, E. Ostuni, D. E. Ingber and G. M. Whitesides, Biomaterials, 1999, 20, 2363–2376 CrossRef CAS PubMed.
- N. Tanaka, H. Ota, K. Fukumori, J. Miyake, M. Yamato and T. Okano, Biomaterials, 2014, 35, 9802–9810 CrossRef CAS PubMed.
- D. Qin, Y. Xia and G. M. Whitesides, Nat. Protoc., 2010, 5, 491–502 CrossRef CAS PubMed.
- H.-W. Chien and W.-B. Tsai, Acta Biomater., 2012, 8, 3678–3686 CrossRef CAS PubMed.
- Z. Wanga, J. Xia, Y. Yan, A.-C. Tsai, Y. Li, T. Maa and J. Guan, Acta Biomater., 2015, 11, 80–87 CrossRef PubMed.
- B. Knöll, C. Weinl, A. Nordheim and F. Bonhoeffer, Nat. Protoc., 2007, 2, 1216–1224 CrossRef PubMed.
- S. Takayama, J. C. McDonald, E. Ostuni, M. N. Liang, P. J. Kenis, R. F. Ismagilov and G. M. Whitesides, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 5545–5548 CrossRef CAS.
- K. Y. Suh, J. Seong, A. Khademhosseini, P. E. Laibinis and R. Langer, Biomaterials, 2004, 25, 557–563 CrossRef CAS PubMed.
- M. C. Berg, S. Y. Yang, P. T. Hammond and M. F. Rubner, Langmuir, 2004, 20, 1362–1368 CrossRef CAS PubMed.
- K. Jang, K. Sato, K. Mawatari, T. Konno and K. Ishihara, Biomaterials, 2009, 30, 1413–1420 CrossRef CAS PubMed.
- K. Itoga, J. Kobayashi, M. Yamato, A. Kikuchi and T. Okano, Biomaterials, 2006, 27, 3005–3009 CrossRef CAS PubMed.
- T. Crouzier, H. Jang, J. Ahn, R. Stocker and K. Ribbeck, Biomacromolecules, 2013, 14, 3010–3016 CrossRef CAS PubMed.
- N. Idota, T. Tsukahara, K. Sato, T. Okano and T. Kitamori, Biomaterials, 2009, 30, 2095–2101 CrossRef CAS PubMed.
- E. A. Roth, T. Xu, M. Das, C. Gregory, J. J. Hickman and T. Boland, Biomaterials, 2004, 25, 3707–3715 CrossRef CAS PubMed.
- S. Iwanaga, Y. Akiyama, A. Kikuchi, M. Yamato, K. Sakai and T. Okano, Biomaterials, 2005, 26, 5395–5404 CrossRef CAS PubMed.
- H. Moriguchi, Y. Wakamoto, Y. Sugio, K. Takahashi, I. Inoue and K. Yasuda, Lab Chip, 2002, 2, 125–132 RSC.
- A. Tourovskaia, T. Barber, B. T. Wickes, D. Hirdes, B. Grin, D. G. Castner, K. E. Healy and A. Folch, Langmuir, 2003, 19, 4754–4764 CrossRef CAS.
- C. H. Cho, J. Park, A. W. Tilles, F. Berthiaume, M. Toner and M. L. Yarmush, BioTechniques, 2010, 48, 47–52 CrossRef CAS PubMed.
- N. Tanaka, H. Moriguchi, A. Sato, T. Kawai, K. Shimba, Y. Jimbo and Y. Tanaka, RSC Adv., 2016, 6, 54754–54762 RSC.
- M. Nishizawa, A. Takahashi, H. Kaji and T. Matsue, Chem. Lett., 2002, 31, 904–905 CrossRef.
- W. Li, Z. Xu, J. Huang, X. Lin, R. Luo, C.-H. Chen and P. Shi, Sci. Rep., 2014, 4, 4784 Search PubMed.
- H. Hisamoto, Y. Nakashima, C. Kitamura, S.-i. Funano, M. Yasuoka, K. Morishima, Y. Kikutani, T. Kitamori and S. Terabe, Anal. Chem., 2004, 76, 3222–3228 CrossRef CAS PubMed.
- M. Mizuhata, T. Miyake, Y. Nomoto and S. Deki, Microelectron. Eng., 2008, 85, 355–364 CrossRef CAS.
- N. Tanaka, H. Ota, K. Fukumori, J. Miyake, M. Yamato and T. Okano, Biomaterials, 2014, 35, 9802–9810 CrossRef CAS PubMed.
- K. Takahashi, K. Mawatari, Y. Sugii, A. Hibara and T. Kitamori, Microfluid. Nanofluid., 2010, 9, 945–953 CrossRef.
- M. Suzuki, A. Kishida, H. Iwata and Y. Ikada, Macromolecules, 1986, 19, 1804–1808 CrossRef CAS.
- Y. K. Kim, H.-H. Ha, J.-S. Lee, X. Bi, Y.-H. Ahn, S. Hajar, J.-J. Lee and Y.-T. Chang, J. Am. Chem. Soc., 2010, 132, 576–579 CrossRef CAS PubMed.
- M. Suzuki, K. Ikeda, M. Yamaguchi, S. N. Kudoh, K. Yokoyama, R. Satoh, D. Ito, M. Nagayama, T. Uchida and K. Gohara, Biomaterials, 2013, 34, 5210–5217 CrossRef CAS PubMed.
- A. Natarajan, M. Stancescu, V. Dhir, C. Armstrong, F. Sommerhage, J. J. Hickman and P. Molnar, Biomaterials, 2011, 32, 4267–4274 CrossRef CAS PubMed.
- Q. Cheng, K. Komvopoulos and S. Li, J. Biomed. Mater. Res., Part A, 2011, 96, 507–512 CrossRef PubMed.
- Y. Tanaka, H. Xi, K. Sato, K. Mawatari, B. Renberg, M. Nilsson and T. Kitamori, Anal. Chem., 2011, 83, 3352–3357 CrossRef CAS PubMed.
- B. Renberg, K. Sato, K. Mawatari, N. Idota, T. Tsukahara and T. Kitamori, Lab Chip, 2009, 9, 1517–1523 RSC.
- K. Shirai, K. Mawatari and T. Kitamori, Small, 2014, 10, 1514–1522 CrossRef CAS PubMed.
- S. H. Kim, S. Iwai, S. Araki, S. Sakakihara, R. Iino and H. Noji, Lab Chip, 2012, 12, 4986–4991 RSC.
- R. Iino, Y. Matsumoto, K. Nishino, A. Yamaguchi and H. Noji, Front. Microbiol., 2013, 4, 300 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra16906f |
| This journal is © The Royal Society of Chemistry 2016 |