
Gold nanocluster formation using morpholino oligomer as template and assembly agent within hybrid bio-nanomaterials†‡
Abstract
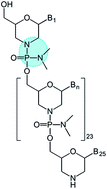
We report the use of a phosphorodiamidate morpholino oligomer as a DNA analogue to template the assembly of a gold nanocluster possessing 6 Au atoms and a diameter of ∼0.9 nm. Spectroscopic characterization indicates that Au preferentially binds to the phosphorodiamidate backbone as opposed to the nucleobases. A high fraction of Au(I) relative to Au(0) is observed by X-ray photoelectron spectroscopy and electrochemistry. This new nanocluster was co-assembled with carbon nanotubes and a fuel-cell enzyme, bilirubin oxidase. The hybrid bio-nanoassembly can reduce O2 to H2O without changing the overpotential of the enzyme.
 Please wait while we load your content...
Please wait while we load your content...

