
NBS-mediated dinitrogen extrusion of diazoacetamides under catalyst-free conditions: practical access to 3-bromooxindole derivatives†
Abstract
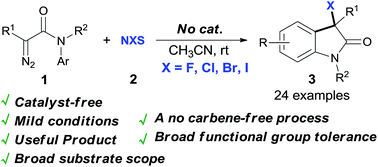
A synthetically useful transformation arises from NBS-mediated dinitrogen extrusion of N-aryl diazoacetamides under catalyst-free conditions, which gives 3-bromooxindoles in high to excellent yields with high selectivity via a non-carbene process.
- This article is part of the themed collection: Elegant Synthetic Routes to Indole Derivatives
 Please wait while we load your content...
Please wait while we load your content...

