
Solvato-morphologically controlled, reversible NIPAAm hydrogel photoactuators†
Aishling Dunne ,
Colm Delaney,
Larisa Florea* and
Dermot Diamond
,
Colm Delaney,
Larisa Florea* and
Dermot Diamond
Insight Centre for Data Analytics, National Centre for Sensor Research, School of Chemical Sciences, Dublin City University, Dublin 9, Glasnevin, Ireland. E-mail: larisa.florea@dcu.ie
First published on 26th August 2016
Abstract
Photo-actuator hydrogels were generated using a N-isopropylacrylamide-co-acrylated spiropyran-co-acrylic acid (p(NIPAAm-co-SP-co-AA)) copolymer, in 100–1–5 mole ratio. Different ratios of deionised water![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :organic solvent (tetrahydrofuran, dioxane and acetone) were used as the polymerisation solvent. By changing the polymerisation solvent, the pore size and density of the hydrogels were altered, which in turn had an impact on the diffusion path-length of water molecules, thus influencing the swelling and photo-induced shrinking kinetics of the hydrogel. We successfully demonstrated that the polymerisation solvent has a significant effect on the curing time, the elasticity and morphology of the resulting hydrogel. The highest shrinking ratio was obtained for hydrogels produced using 4:1 acetone:deionised water (CI) as the polymerisation solvent, with the hydrogel reaching 39.56% (±2.37% (n = 3)) of its hydrated area after 4 min of white light irradiation followed by reswelling in the dark to 61.95% (±5.76% (n = 3)) after 11 min. Conversely, the best reswelling capabilities were obtained for the hydrogels produced using 1:1 tetrahydrofuran:deionised water (AIII), when the shrunk hydrogel (61.78 ± 0.26% (n = 3)) regained 91.31% (±0.22% (n = 3)) of its original size after 11 min in the dark. To our knowledge, this is the largest reported photo-induced area change for self-protonated spiropyran containing hydrogels. The shrinking/reswelling process was completely reversible in DI water with no detectable hysteresis over three repeat irradiation cycles.
:organic solvent (tetrahydrofuran, dioxane and acetone) were used as the polymerisation solvent. By changing the polymerisation solvent, the pore size and density of the hydrogels were altered, which in turn had an impact on the diffusion path-length of water molecules, thus influencing the swelling and photo-induced shrinking kinetics of the hydrogel. We successfully demonstrated that the polymerisation solvent has a significant effect on the curing time, the elasticity and morphology of the resulting hydrogel. The highest shrinking ratio was obtained for hydrogels produced using 4:1 acetone:deionised water (CI) as the polymerisation solvent, with the hydrogel reaching 39.56% (±2.37% (n = 3)) of its hydrated area after 4 min of white light irradiation followed by reswelling in the dark to 61.95% (±5.76% (n = 3)) after 11 min. Conversely, the best reswelling capabilities were obtained for the hydrogels produced using 1:1 tetrahydrofuran:deionised water (AIII), when the shrunk hydrogel (61.78 ± 0.26% (n = 3)) regained 91.31% (±0.22% (n = 3)) of its original size after 11 min in the dark. To our knowledge, this is the largest reported photo-induced area change for self-protonated spiropyran containing hydrogels. The shrinking/reswelling process was completely reversible in DI water with no detectable hysteresis over three repeat irradiation cycles.
1. Introduction
Hydrogels can be defined as three dimensional polymer matrices which are able to hold large quantities of water in relation to their size.1 In recent years hydrogels have been used in a number of different applications such as diagnostics and therapeutics,2,3 tissue engineering,4 wound dressings5 and drug delivery,6–8 microfluidics,9 among others.10,11 Stimuli-responsive hydrogels in particular have gained increasing attention over the last decade due to their responsive nature towards internal or external stimuli, with little or no human intervention.9,12 By incorporating stimuli-responsive units in their structure, hydrogel actuators can be developed, that respond to a variety of stimuli such as light,10,11,13,14 pH,10,15,16 electric10,11 or magnetic fields10,11,17 and temperature.10,11,15,18 Amongst these, photo-responsive materials are particularly attractive as they can be actuated externally, in a non-invasive, non-contact manner.In recent years, photo-responsive hydrogels reported in the literature have been overwhelmingly focused on crosslinked copolymers of N-isopropylacrylamide (NIPAAm) with spiropyran photochromic derivatives. One disadvantage of this approach was the need for an external acidic environment (typically 10 mM HCl) to ensure protonation of the spiropyran (SP) to the protonated merocyanine (MC-H+) form. The MC-H+ form is more hydrophilic compared to the SP form, and its formation triggers swelling of the gel. When irradiated with white/blue light (λmax MC-H+ = 422 nm),19,20 the MC-H+ form is converted back to the more hydrophobic SP form and the free protons diffuse into the external environment. Simultaneously, the polymer network collapses into the compact globular form, the water is expelled, and the hydrogel contracts. In order to induce re-swelling, the hydrogel had to be immersed again in acidic environments to drive formation of the protonated merocyanine.14,20,21 Additionally, the re-swelling times were long, typically up to several hours.14,19,20 These disadvantages restricted p(NIPAAm-co-SP) photo-actuated hydrogels to single-use applications.
Recently, we reported that the addition of acrylic acid (AA), which, when copolymerised within the hydrogel, provides an internal source of protons for reversible hydrogel photo-actuation in neutral pH environments.22,23 The addition of the AA comonomer successfully removed the requirement for external acidic conditions and expanded the working range of the photo-actuator to pH 2–7, making it suitable for a wide variety of biological applications. However, the prolonged shrinking and reswelling times (20 min of white light irradiation to reach ∼82% of its fully hydrated size followed by 60 min in the dark to achieve reswelling up to 98%),22 constituted significant limitations for application of these photo-actuators.
In order to improve the shrinking/swelling kinetics of hydrogels, several approaches have been undertaken including (1) the introduction of novel materials, such as ionic liquids, to tune the overall hydrophilicity/hydrophobicity of the gel network,24–26 (2) the use of pore-forming agents to create highly porous gels with shorter average path lengths for water diffusion,27,28 (3) variation of the photochromic unit itself, through the use of electron withdrawing or electron-donating substituents as means to control the ring-opening rates20,21 and (4) varying polymerisation solvents mixtures to influence the morphology of hydrogels by directly effecting pore generation during polymerisation.29–32
In this study, photo-actuator hydrogels were generated using a N-isopropylacrylamide-co-acrylated spiropyran-co-acrylic acid (p(NIPAAm-co-SP-co-AA)) copolymer, in a 100–1–5 mole ratio. In order to allow for control of the photo-induced shrinking and reswelling times of the hydrogels, different ratios of DI water:organic solvent (tetrahydrofuran (THF), dioxane and acetone) were used as the polymerisation solvent. Varying the organic solvent, and the volume ratio of the solvent mixtures resulted in hydrogels with varying porosity, which in turn has affected the physical and mechanical properties, swelling/shrinking degree and photo-actuation kinetics of the hydrogel system.
2. Experimental
2.1 Materials
N-Isopropylacrylamide 98% (NIPAAm), N,N′-methylenebisacrylamide 99% (MBIS), phenylbis(2,4,6 trimethyl benzoyl)phosphine oxide 97% (PBPO), acrylic acid (180–200 ppm MEHQ as inhibitor) 99% (AA), tetrahydrofuran 99% (THF), 1,4-dioxane 99% (dioxane), acetone 99%, anhydrous dichloromethane (50–150 ppm amylene as stabilizer) 99% (DCM), ethyl acetate 99%, n-hexane 95%, were acquired from Sigma Aldrich, Ireland and used as received.1′,3′,3′-Trimethyl-6-acryloylspiro(2H-1-benzopyran-2,2-indoline) (SP-A) was synthesised as described elsewhere.22
2.2 Single-crystal X-ray diffraction
Crystals of SP-A, suitable for single-crystal X-ray diffraction, were grown by slow diffusion from hexane:ethyl acetate (8:1). The structure was solved and refined using the Bruker SHELXTL Software Package, with a P21/c space group, for Z = 4. The final anisotropic full-matrix least-squares refinement on F2 with 238 variables converged at R1 = 6.29%, for the observed data and wR2 = 17.36% for all data. The goodness-of-fit was 1.029. The molecules were observed to pack in a head-to-tail manner, as shown in Fig. S1,† with strong π–π interactions between adjacent benzopyran and indoline portions of neighbouring molecules.
2.3 Gel preparation
For hydrogel synthesis, the monomeric cocktail consisted of 200 mg NIPAAm, 8.17 mg MBIS (3 mol% relative to NIPAAm), 6.09 mg SP-A (1 mol% relative to NIPAAm), 7.34 mg PBPO (1 mol% relative to NIPAAM) and 6.03 μL AA (5 mol% relative to NIPAAm) dissolved in 500 μL of the polymerisation solvent (4:1, 2:1 and 1:1, respectively, vol/vol, organic solvent:DI water). Circular hydrogels were prepared in a home-made cell consisting of a 1H,1H,2H,2H-perfluorodecyltriethoxy saline functionalised glass slide and a glass cover slide separated by a 250 μm high spacer made out of poly(methyl methacrylate)/pressure sensitive adhesive (PMMA/PSA) (Fig. S2†). The cell was filled by capillary action with the monomer solution and subsequently exposed to white light through a photo-mask consisting of 1 mm diameter transparent disks. The polymerisation time was 20–30 s, depending on the polymerisation solvent mixture employed. The white light source used was a Dolan-Jenner-Industries Fiber-Lite LMI LED lamp having two gooseneck waveguides placed at a distance range of 1 to 2 cm from the platform. The light intensity measured with a Multicomp LX-1309 light meter was 320–337 klux. After polymerisation, the obtained circular hydrogels were washed gently with ethanol and DI water to remove any unpolymerised materials and allowed to swell in deionised water for 4–6 hours to ensure full hydration.
2.4 Rheology
Rheology curing measurements were performed on the unpolymerised cocktail. The measurements were carried out on an Anton Paar MCR 301 rheometer using a CP50-2 tool with a diameter of 49.97 mm and a cone angle of 1.996°. White light curing was initiated after 60 s with a light intensity of 200–225 klux at 25 °C. Rheology studies of the hydrated gels were done using the PP15 parallel plate tool of 15 mm diameter. For this, amplitude sweep tests were carried out at a 100 rad s−1 angular frequency and a normal force of 1 N.2.5 Scanning electron microscopy
The hydrogel samples were first swollen in DI water, and then frozen with liquid nitrogen and subsequently freeze-dried using a Labconco freeze-drier, model 7750060. The samples were kept overnight at 0.035 mbar pressure and a temperature of −40 °C. The freeze-dried hydrogels were imaged using Scanning Electron Microscopy (SEM) performed on a Carl Zeiss EVOLS 15 system at an accelerating voltage of 14.64 kV. Samples were placed onto silicon wafers and coated with a 10 nm gold layer prior to imaging.2.6 Photo-actuation measurements
For white light irradiation shrinking and reswelling measurements, the hydrogels were placed in a custom-made cell, comprising 2 cover glass slides separated by 500 μm high spacers made out of PMMA/PSA. The imaging was done with an Aigo GE-5 microscope using a 60× objective lens with the accompanying software. The light was provided by a Dolan-Jenner-Industrie Fiber-Lite LMI with a light intensity of ∼210 klux through two waveguide goosenecks placed 2–3 cm away from the sample. Each sample was exposed to white light irradiation for 4 min then placed in the dark for a further 11 min. The area measurements of the freestanding hydrogel discs were performed using Image J (1.47v) software. Three different hydrogels were measured for each point and the relative area% was calculated using the following equation (n = 3):At = measured area at time t; Ao = area of a fully hydrated gel.
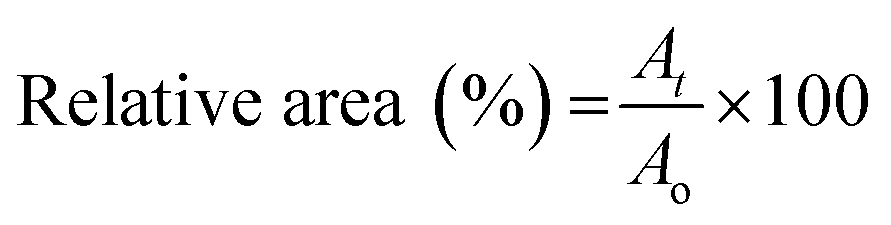
3. Results and discussion
3.1 Photo-induced curing
Rheology was used to study the effects of the polymerisation solvent on the curing times and properties of the hydrogels obtained. The photo-induced curing measurements were carried out for each monomeric cocktail using different polymerisation solvent mixtures, as described in the Experimental section. All samples were exposed to white light and the storage modulus recorded. The storage modulus increases with time and in most cases (AI, AII, AIII, BI, BII and CI) reaches a plateau indicating no further significant growth of elastic structures due to the crosslinking.33 It was observed that the plateau region of the storage modulus was dependent on the polymerisation solvent used (Fig. 1 and S4–S6†). For the 4:1 organic solvent:DI water series, the storage moduli remain relatively unchanged after ∼190 s of white light irradiation (Fig. 1). The hydrogels which were polymerised in the presence of dioxane:DI water showed the highest storage modulus plateau (Fig. S4†), suggesting that when dioxane was used in the polymerisation mixture, hydrogels with greater elastic properties were produced (Table 1).34
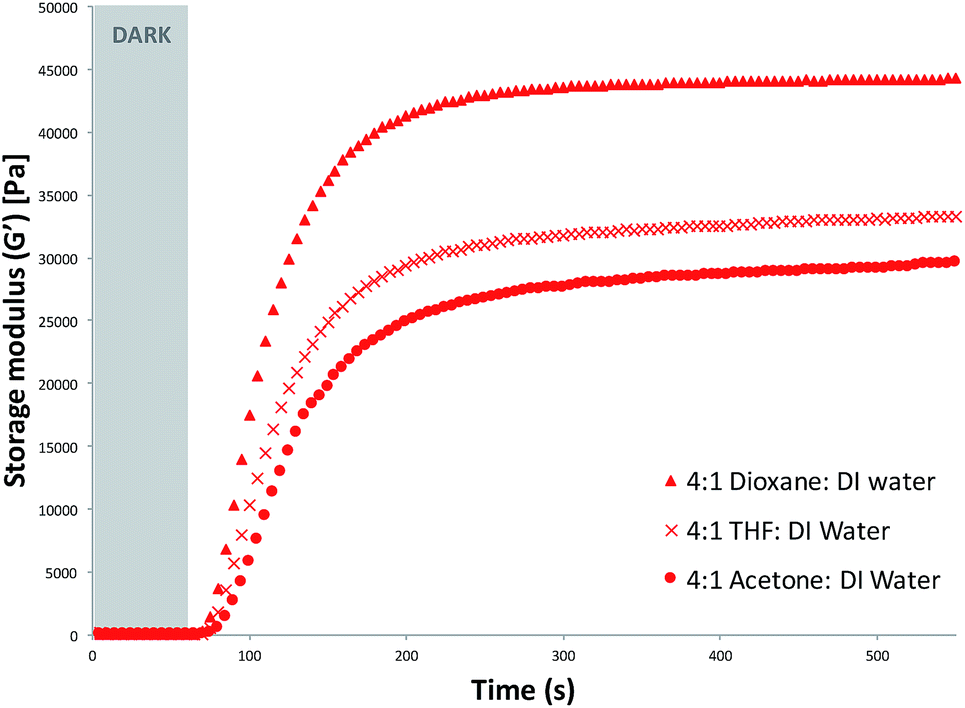 | ||
| Fig. 1 Photo-curing of hydrogels produced when the polymerisation solvent was 4:1 (v/v) organic solvent (dioxane, THF and acetone, respectively):deionised water. White light polymerisation was initiated after 60 s. | ||
| Gel ID | Polymerisation solvent | G′ [Pa] | G′′ [Pa] | tanδ (G′′/G′) |
Gel point [s] |
|---|---|---|---|---|---|
| AI | 4:1 THF:DI water |
32600 |
168 | 5.15 × 10−3 | 70 |
| AII | 2:1 THF:DI water |
30700 |
110 | 3.58 × 10−3 | 70 |
| AIII | 1:1 THF:DI water |
12000 |
184 | 1.53 × 10−2 | 70 |
| BI | 4:1 dioxane:DI water |
44000 |
74.9 | 1.70 × 10−3 | 70 |
| BII | 2:1 dioxane:DI water |
44400 |
49.8 | 1.12 × 10−3 | 70 |
| BIII | 1:1 dioxane:DI water |
47700 |
202 | 4.23 × 10−3 | 75 |
| CI | 4:1 acetone:DI water |
28700 |
30.2 | 1.05 × 10−3 | 70 |
| CII | 2:1 acetone:DI water |
35900 |
93.8 | 2.61 × 10−3 | 70 |
| CIII | 1:1 acetone:DI water |
13000 |
80.5 | 6.19 × 10−3 | 75 |
During the curing process the storage modulus quickly surpassed the loss modulus. This is known as the “gel point” and is where the material exhibits more elastic than viscous behaviour. For all the samples examined, the gel point was achieved within the first ∼70 s after the start of the experiment, meaning that it was reached within ∼10 s after the photo-polymerisation was initiated (white light is turned on after 60 s).
tanδ is a representation of the ratio of the elastic (storage modulus) and viscous (loss modulus) moduli, (G′′/G′). This parameter helps to identify how tacky/sticky the hydrogel is (Table 1). It was observed that hydrogels which were polymerised in the presence of 1:1 organic solvent:DI water had the highest tanδ values in their respective series (Table 1). These results suggest that using lower organic solvent content in the polymerisation mixture produced hydrogels which were tackier/stickier and more delicate to handle.
3.2 Hydrogel morphology
A Scanning Electron Microscopy (SEM) study was carried out as described in the Experimental section, to investigate the effect of different polymerisation solvents on the morphology of the resulting hydrogels. Varying solvent ratios was seen to produce hydrogels of different pore sizes. For example, when THF:DI water was employed as the polymerisation solvent (Fig. 2), it was revealed that as the organic solvent content was decreased, the pore size also decreased. This is possibly due to the well-known co-nonsolvency of pNIPAAm in different solvent mixtures, including THF:water where, depending on the solvent ratio, pNIPAAm shows drastic conformation changes from a fully swollen coil to a globular state.32,35–37 For example, in the case of MeOH:water mixtures, it has been shown that pNIPAAm becomes insoluble at a molar ratio of MeOH (XMeOH) between 0.13–0.4, while for XMeOH < 0.13 and XMeOH > 0.4 pNIPAAm is soluble.35 Similar observations have been reported for other binary solvent systems such as THF:water, DMSO:water and acetone:water, for which the solubility of pNIPAAm depends on the ratio of the two solvents.32,37,38 It is therefore believed that as the polymeric chains are growing they will either precipitate in a globular conformation or adopt an extended coil conformation due to their solvency. This in turn determines the pore size and density of the resulting hydrogel. From the representative SEM images of the THF:DI water series, it is revealed that the pore size range of the hydrogels polymerised in the presence of 1:1 v/v THF:DI water (AIII) was 1.11–1.66 μm (Fig. 2c), considerably smaller when compared to 4:1 volume ratio (AI), which resulted in hydrogels having pore sizes ranging from 5.61–6.47 μm (Fig. 2a). This shows the direct effect of the solvent ratio on the pore size development. The hydrogels which were polymerised in the presence of dioxane:DI water did not follow the pore development pattern seen for THF:DI water, but produced gels with a much broader pore size distribution in the range of 4.75 μm to 8.15 μm (Fig. S6–S8†).
 | ||
| Fig. 2 SEM images of hydrogels synthesised using different (v/v) ratios of THF:DI water as the polymerisation solvent. | ||
3.3 Oscillation analysis
Amplitude sweep measurements (Fig. 3 and ESI Fig. S12 and S13†) were performed on the fully hydrated gels in order to determine their mechanical properties and relate these with the porosity of the hydrogels. It would be expected that the hydrogels with larger pore sizes would show weaker mechanical properties due to less polymer being present. This correlation was evident when comparing the results from the oscillation study of the hydrated gels with the SEM data. In the case when THF:DI water mixture was used as the polymerisation solvent, amplitude sweep measurements (Fig. 3) show that the AIII and AII hydrogels have a higher (visco)elastic modulus (10–13 kPa) compared to the AI hydrogel (∼5 kPa). Intuitively, one would expect that highly porous gels are more flexible, less stiff and cannot withstand high stress, causing them to reach the linear viscoelastic range (LVE) quicker than less porous polymers. In this study we have selected 10% as tolerance threshold and all G′ values below 90% of the plateau value are considered to be outside the LVE range. In this context, the AIII hydrogel can withstand higher shear stress before it reaches the linear viscoelastic range (LVE) of ∼1 kPa. The AI and AII hydrogels reached the LVE limit quicker and with less shear stress of ∼300 Pa.
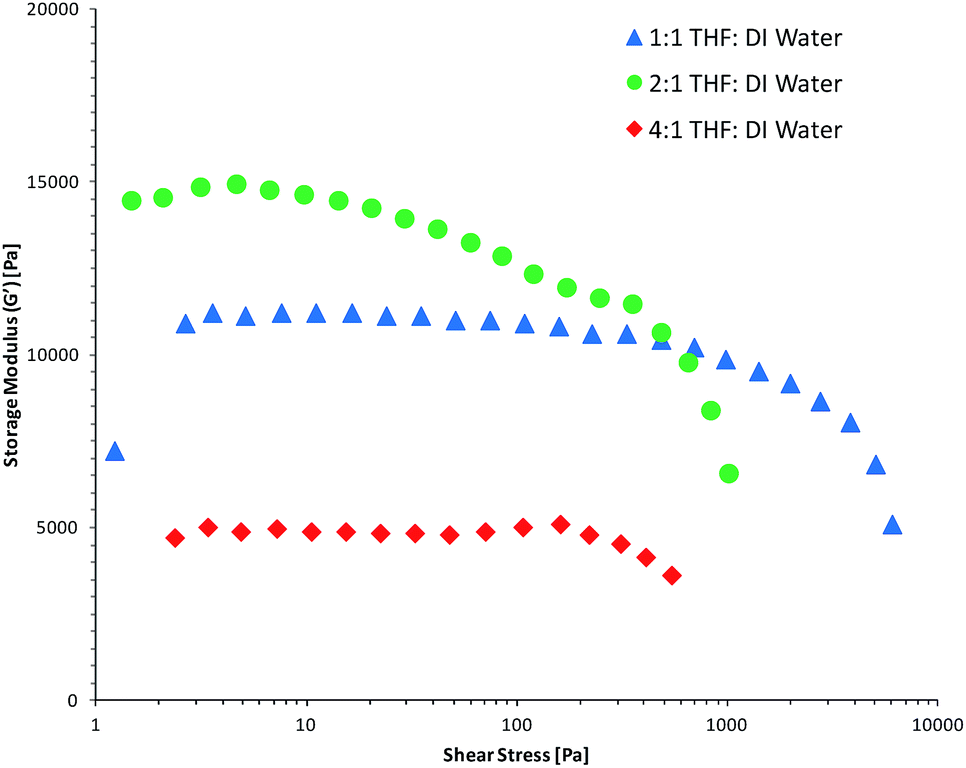 | ||
| Fig. 3 Storage moduli versus shear stress of the hydrated hydrogels polymerised in the presence of THF:DI water solvent mixtures during a strain amplitude sweep using a normal force of 1 N. | ||
The amplitude sweep results for the hydrogels with acetone:DI water and dioxane:DI water as the polymerisation solvents (ESI Fig. S12 and S13,† respectively) appear to correlate with the pore sizes obtained from the SEM study (ESI Fig. S6–S11†). In the case of dioxane:water the pore density, curing studies and the strain amplitude sweeps obtained for all the different solvent ratios have given very similar results. Moreover, the LVE limits for all of the B gels (BI–BIII) have similar values of ∼800 Pa, showing that the B gels with similar porosity reach the linear viscoelastic range under similar conditions. Analogous comparisons can be made for hydrogels polymerised in the presence of acetone:DI water (LVE range 1–2.2 kPa).
Overall the increase in hydrogel porosity decreases the stiffness and mechanical strength of the hydrogels. Nevertheless, all the hydrogels obtained in this study are still strong enough to be handled for actuation and measuring purposes, and posses storage modulus values of about 5–20 kPa.39
3.4 Photo-actuation study
The main objective of this study was the correlation of the photo-induced shrinking and subsequent swelling of the p(NIPAAm-co-SP-co-AA) hydrogels in relation to their solvato-induced porosity and mechanical properties. In this context, a photo-actuation study was carried out on the nine hydrogel variations polymerised in the presence of different solvent mixtures. The expansion and contraction of each hydrogel sample were measured in a series of three actuation cycles in DI water in order to investigate the reversibility of photo-actuation. When the p(NIPAAm-co-SP-co-AA) hydrogels were immersed in DI water, in the dark, they expanded due to the protonation of the SP unit to the more hydrophilic MC-H+ by the AA comonomer (pKa ∼ 4.5) which serves as an internal acid source (Fig. 4). Under these conditions the hydrogel has a yellow colour due to the absorption spectrum of MC-H+ (λmax = 422 nm).19 When the same hydrogel was irradiated with white light, a rapid colour change was observed due to the deprotonation of MC-H+ and its reversion back to the colourless SP form. The SP form is more hydrophobic causing the polymer chains to collapse, thereby expelling water and triggering hydrogel contraction.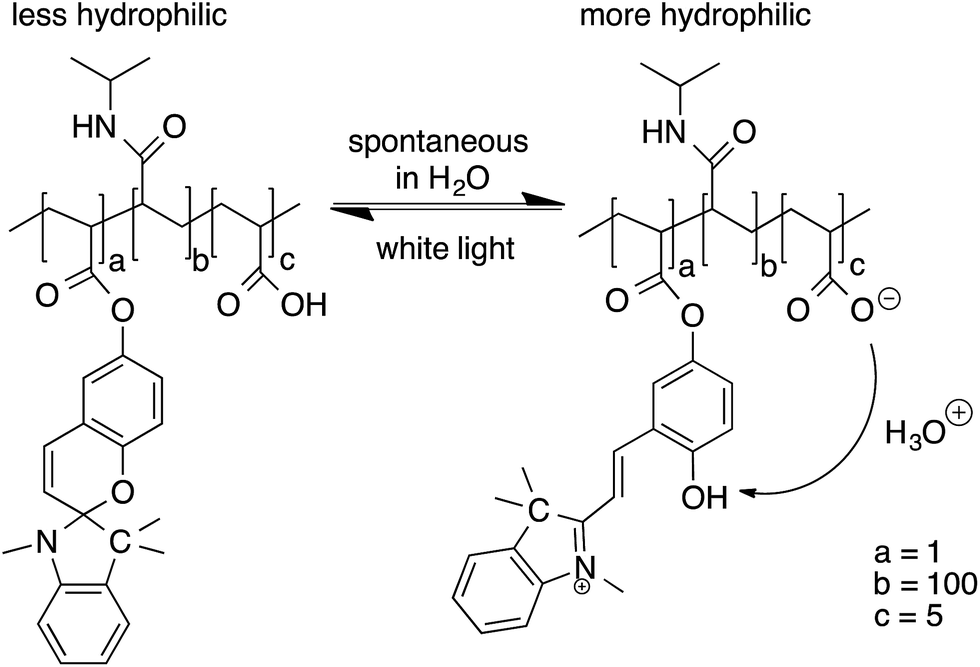 | ||
| Fig. 4 Chemical structure of p(NIPAAm-co-SP-co-AA) polymer chains under different illumination conditions. | ||
During this study, the fully hydrated gels were exposed to white light for 4 min and returned to the dark for a further 11 min to allow reswelling and this process was repeated three times (Fig. 5, 6 and S14–S20†). These times were sufficient to reach a close steady state (Fig. S21–S23†). The area contraction and expansion of the hydrogels polymerised in the presence of THF:DI water are shown in Fig. 5. Comparing AI, AII and AIII a difference in the degree of hydrogel contraction was observed. The hydrogel sample AI have contracted the most in this series (to 51.43% of their initial size), when compared to the hydrogels AII (58.94%) and AIII (61.78%) (Table 2). The results are in agreement with the pore sizes obtained, where the AI hydrogels showed the largest pores of the series, enhancing the degree of shrinking of this sample by improving the water diffusion out of the hydrogel. However, the expansion in relative area for the AII hydrogel is much lower (75.17%) than the AI and AIII hydrogels (82.99% and 91.31% respectively). This may be attributed to the increased storage modulus of the AII (Fig. 3) which can contribute to the poor reswelling of the gel and thus the decreased relative area change.
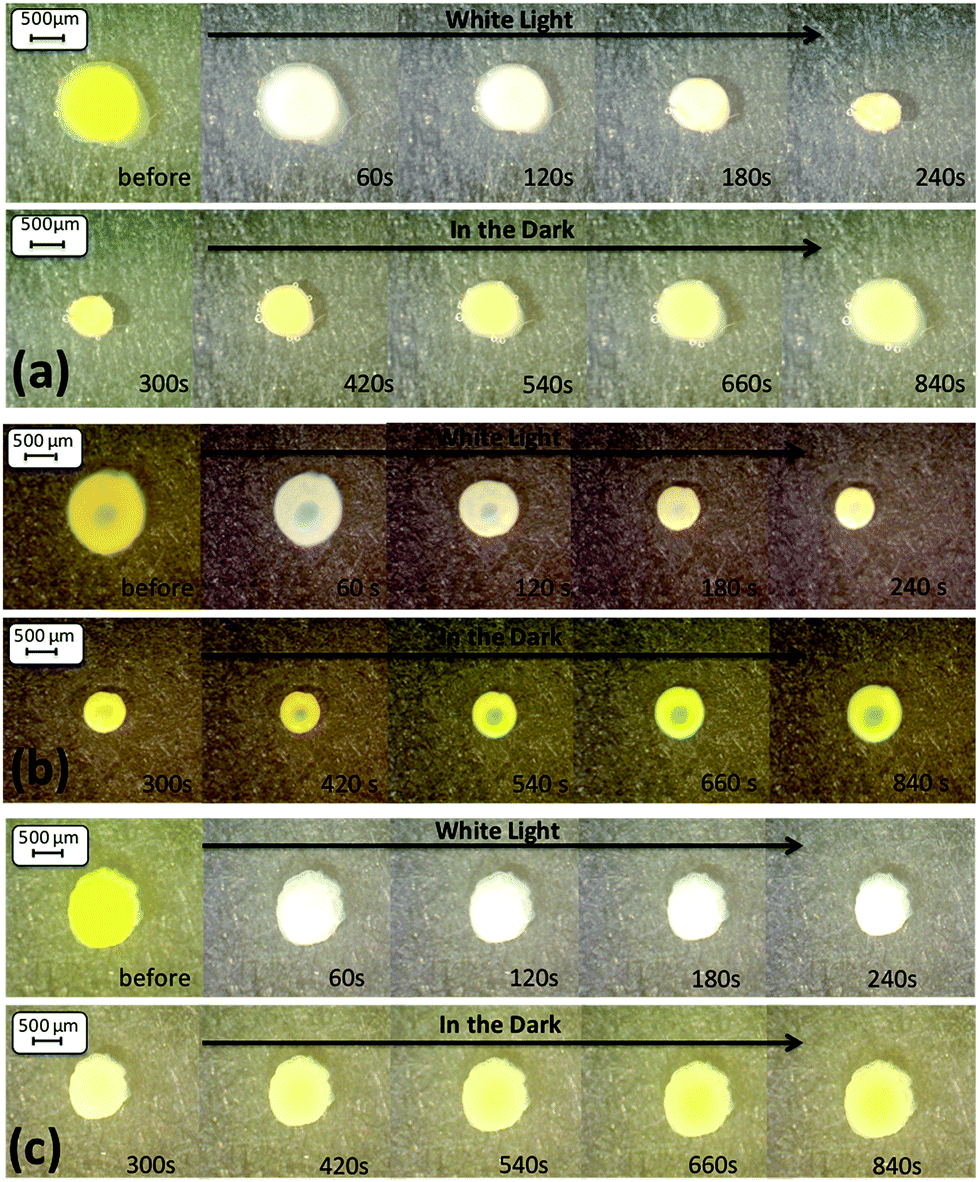 | ||
| Fig. 5 Microscope images (magnification ×60) of the hydrogel shrinking and reswelling area of the three v/v ratios ((a) AI, (b) AII and (c) AIII) of the THF:DI water solvent mixture. | ||
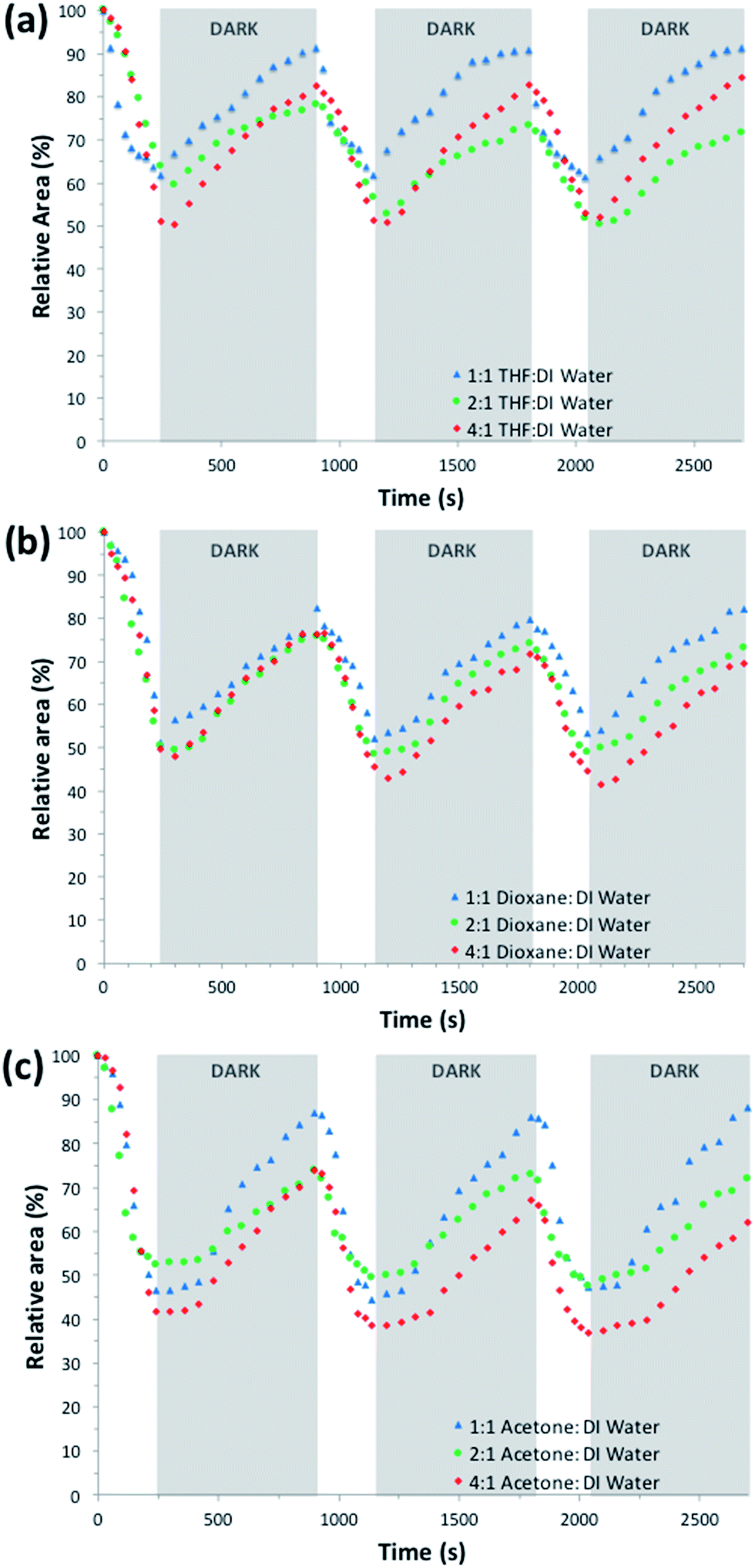 | ||
| Fig. 6 Photo-actuation cycles of hydrogel samples produced in the presence of different polymerisation solvent mixtures ((a) THF:DI water, (b) dioxane:DI water, (c) acetone:DI water), in real time. | ||
| Gel ID | Relative area [%] after contraction | Error [%] (n = 3) after contraction | Relative area [%] after expansion | Error [%] (n = 3) after expansion | Relative area change [%] |
|---|---|---|---|---|---|
| AI | 51.43 | 0.95 | 82.99 | 0.87 | 31.64 |
| AII | 58.94 | 6.04 | 75.17 | 3.39 | 16.23 |
| AIII | 61.78 | 0.26 | 91.31 | 0.22 | 29.53 |
| BI | 47.23 | 2.62 | 73.36 | 3.44 | 26.13 |
| BII | 49.51 | 1.08 | 74.65 | 1.37 | 25.14 |
| BIII | 51.83 | 1.01 | 81.57 | 1.36 | 29.74 |
| CI | 39.56 | 2.37 | 61.95 | 5.76 | 22.39 |
| CII | 50.34 | 2.35 | 71.63 | 3.26 | 21.29 |
| CIII | 46.07 | 1.30 | 86.95 | 0.92 | 40.88 |
In the case of dioxane:DI water as the polymerisation solvent mixture (Fig. S14 (BI), S15 (BII) and S16 (BIII)†), there is not a clear difference between the contraction values (47.23%, 49.51%, and 51.83% respectively) or the expansion values (73.36%, 74.65%, and 81.57% respectively) (Table 2). These results would be expected, taking into account their similar pore sizes (Fig. S6–S8†) and mechanical properties (G′ = 44–47 kPa, Fig. S4†).
In the acetone:DI water series, the CI hydrogel shows the highest relative shrinking (hydrogels shrink to 39.56% of their hydrated size) but on reswelling it only reaches 61.95% of its initial size. In contrary, the CIII hydrogel shows a smaller relative shrinking to 46.07% (Table 2), but over all shows the highest relative area change upon repeating actuation cycles (40.88%). This could be attributed to its high pore density of various sizes (ESI Fig. S11†) compared to the other ratios, which in turn has been shown to improve water diffusion in the hydrogel. For each of the polymerisation solvent mixtures, the photo-induced contraction and expansion of the hydrogels were shown to be successfully reproducible over at least three illumination cycles with minimal hysteresis (Fig. 6).
During this study the polymerisation solvent mixture and volume ratio were shown to play a crucial role in the resulting morphology and mechanical properties of the hydrogels. The THF:DI water mixture best highlighted the importance of the polymerisation solvent. During the curing study the AI hydrogel had the highest storage modulus value of 32600 Pa, which resulted in hydrogels that were robust and easy to handle. Upon swelling, however, the mechanical properties of the gel drastically changed, showing the smallest storage modulus of the series, at 5000 Pa. This is the first indication that the AI hydrogel absorbed the highest amount of water of the series, as indicated also by its high porosity. In comparison, the highest tanδ value of the series, of 1.55 × 10−2 was observed for the AIII hydrogel, resulting in a tacky hydrogel with smaller pores (1.66–1.11 μm), that upon water absorption had a much higher storage modulus of ∼12 kPa, indicating a hydrogel with higher mechanical strength compared to AI hydrogel. Furthermore, the higher storage modulus of the hydrated AII hydrogel (∼15 kPa) implies an even more rigid material, making it less susceptible to photo-induced actuation (Table 2). This demonstrates that using polymerisation solvents with different ratios of organic solvents can dramatically affect the mechanical properties of the polymerised hydrogel due to the relative solvency of the growing polymeric chains (mainly composed of pNIPAAm) in the solvent mixture. As described above, there is a direct correlation between the solvent used during polymerisation, the morphology of the resulted hydrogels and the photo-actuation degree. Impressive variations in the overall photo-induced area change of p(NIPAAm-co-SP-co-AA) hydrogels from ∼15–40% can be observed (Table 2) just by changing the polymerisation solvent mixture. This study provides essential information on the importance of the polymerisation environment in allowing the synthesis of pNIPAAm-based hydrogels with desired actuation responses.
4. Conclusions
In this study, nine different variations of organic solvent:DI water mixtures were used as polymerisation solvents to produce photo-actuating p(NIPAAm-co-SPA-co-AA) hydrogels, with the aim to control their morphology and therefore their actuation capabilities. The work has successfully demonstrated how the polymerisation solvent affects the morphology of the resulting hydrogel, directly influencing the curing properties, pore sizes, physical properties and degree of photo-actuation. This development has allowed for greater degrees of photo-actuation than previously reported and provides critical information for the development of micro-valves and photo-regulated flow controllers of optimal response times, in microfluidic devices.
Acknowledgements
This project has been funded by Science Foundation Ireland under the Insight initiative, grant SFI/12/RC/2289. LF and DD acknowledge COST Action MP1205 – ‘Advances in Optofluidics: integration of optical control and photonics with microfluidics’. The authors thank Dr Bartosz Ziółkowski for helpful discussions on the rheology data.Notes and references
- N. A. Peppas and A. R. Khare, Adv. Drug Delivery Rev., 1993, 11, 1–35 CrossRef CAS.
- J. M. Rosiak and F. Yoshii, Nucl. Instrum. Methods Phys. Res., 1999, 151, 56–64 CrossRef CAS.
- Y. Tabata and T. Asahara, US Pat., App 11884, 2006.
- K. Lee and D. J. Mooney, Chem. Rev., 2001, 101, 1869–1880 CrossRef CAS PubMed.
- B. Balakrishnan, M. Mohanty and P. R. Umashankar, Biomaterials, 2005, 26, 6335–6342 CrossRef CAS PubMed.
- B. Kim and Y. Shin, J. Appl. Polym. Sci., 2007, 105, 3656–3661 CrossRef CAS.
- T. R. Hoare and D. S. Kohane, Polymer, 2008, 49, 1993–2007 CrossRef CAS.
- N. A. Peppas, Curr. Opin. Colloid Interface Sci., 1997, 2, 531–537 CrossRef CAS.
- D. Liang and J. Hongrui, Soft Matter, 2007, 3, 1223–1230 RSC.
- A. Suk-kyun, M. K. Rajeswari, K. Seong-Cheol, S. Nitin and Z. Yuxiang, Soft Matter, 2008, 4, 1151–1157 RSC.
- H. Meng and H. Jinlian, J. Intell. Mater. Syst. Struct., 2010, 21, 859–885 CrossRef CAS.
- S. Shinji, S. Kimio, O. Katsuhide, H. Kazuaki, T. Toshiyuki and K. Toshiyuki, Sens. Actuators, A, 2007, 140, 176–184 CrossRef.
- S. Kimio, T. Toshiyuki, S. Taku and K. Toshiyuki, J. Photochem. Photobiol., A, 2013, 261, 46–52 CrossRef PubMed.
- K. Sumaru, K. Ohi, T. Takagi, T. Kanamori and T. Shinbo, Langmuir, 2006, 22, 4353–4356 CrossRef CAS PubMed.
- X. Gao, Y. Cao, X. Song, Z. Zhang, C. Xiao, C. He and X. Chen, J. Mater. Chem. B, 2013, 1, 5578 RSC.
- N. Sina, M. R. Joselito, G. W. Philip, G. W. Gordon and M. S. Geoffrey, J. Polym. Sci., Part B: Polym. Phys., 2012, 50, 423–430 Search PubMed.
- M. Zrínyi, Colloid Polym. Sci., 2000, 278, 98–103 Search PubMed.
- B. Ziółkowski and D. Diamond, Chem. Commun., 2013, 49, 10308–10310 RSC.
- S. Kimio, K. Mitsuyoshi, K. Toshiyuki and S. Toshio, Macromolecules, 2004, 37, 4949–4955 CrossRef.
- T. Satoh, K. Sumaru, T. Takagi, K. Takai and T. Kanamori, PCCP Phys. Chem. Chem. Phys., 2011, 13, 7322–7329 RSC.
- S. Taku, S. Kimio, T. Toshiyuki and K. Toshiyuki, Soft Matter, 2011, 7, 8030–8034 RSC.
- B. Ziółkowski, L. Florea, J. Theobald, F. Benito-Lopez and D. Diamond, Soft Matter, 2013, 9, 8754–8760 RSC.
- J. E. Stumpel, B. Kowski, L. Florea, D. Diamond, D. J. Broer and A. Schenning, ACS Appl. Mater. Interfaces, 2014, 6, 7268–7274 CAS.
- S. Gallagher, A. Kavanagh, B. Zíołkowski, L. Florea, D.R. MacFarlane, K. Fraser and D. Diamond, Phys. Chem. Chem. Phys., 2014, 16, 3610–3616 RSC.
- J. Le Bideau, L. Viau and A. Vioux, Chem. Soc. Rev., 2011, 40, 907–925 RSC.
- F. Benito-Lopez, R. Byrne, A. Răduţă, N. Vrana, G. McGuinness and D. Diamond, Lab Chip, 2010, 10, 195–201 RSC.
- T. Singh, M. J. Garland, K. Migalska, E. Salvador, R. Shaikh, H. O. McCarthy, D. A. Woolfson and R. F. Donnelly, J. Appl. Polym. Sci., 2012, 125, 2680–2694 CrossRef CAS.
- B. Ziółkowski, L. Florea, J. Theobald, F. Benito-Lopez and D. Diamond, J. Mater. Sci., 2016, 51, 1392–1399 CrossRef.
- X. Zhang, R. Zhuo and Y. Yang, Biomaterials, 2002, 23, 1313–1318 CrossRef CAS PubMed.
- G. Filipcsei, S. Kimio, T. Toshiyuki, K. Toshiyuki and Z. Miklós, J. Mol. Liq., 2014, 189, 63–67 CrossRef.
- H. M. Crowther and B. Vincent, Colloid Polym. Sci., 1998, 276, 46–51 CAS.
- M. W. Francoise, H. Ringsdorf and J. Venzmer, Macromolecules, 1990, 23, 2415–2416 CrossRef.
- Y. Wang, D. A. Lakho and D. Yao, J. Compos. Mater., 2015, 67, 132–134 Search PubMed.
- T. G. Mezger, 114–143.
- I. Bischofberger, D. C. E. Calzolari and V. Trappe, Soft Matter, 2014, 1–8 CAS.
- J. Hao, H. Cheng, P. Butler, L. Zhang and C. C. Han, J. Chem. Phys., 2010, 132, 154902 CrossRef PubMed.
- H. G. Schild, M. Muthukumar and D. A. Tirrell, Macromolecules, 1991, 24, 948–952 CrossRef CAS.
- R. O. Costa and R. F. Freitas, Polymer, 2002, 43, 5879–5885 CrossRef CAS.
- K. S. Anseth, C. N. Bowman and L. Brannon-peppas, Biomaterials, 1996, 17, 1647–1657 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1475017. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra16807h |
| This journal is © The Royal Society of Chemistry 2016 |