DOI:
10.1039/C6RA16797G
(Paper)
RSC Adv., 2016,
6, 71025-71045
Tweaking of the supramolecular gelation properties of a dipeptide based ambidextrous organogelator through the cooperative influence of hydrophobicity, steric bulk and conformational flexibility of the side chain residue of a single hydrophobic α-amino acid encrypted on a designed molecular frame†
Received
29th June 2016
, Accepted 12th July 2016
First published on 12th July 2016
Abstract
The cooperative influence of the side chain hydrophobicity, steric volume and conformational flexibility of a single hydrophobic α-amino acid (αAA) has been found to be pivotal in the tuning of the gelation properties of a dipeptide based ambidextrous organogelator, N-n-hexadecanoyl-αAA-(β)Ala-OBn [αAA = L-isomers of Ala (4a), Val (4b), Leu (4c), Ile (4d), Phe (4e) and Gly (4f)]. An optimal balance among all the above parameters becomes apparently ideal for 4b ≥ 4d > 4a > 4c, whereas, between 4c and 4d with isomeric side chains, 4c shows much weaker gelation. Moreover, 4e gets the extra stability due to its side chain π–π type attractive interaction with an apparent pseudocentro-symmetric H-bonded amide network. The aromatic unit at the C-termini apparently helps a parallel β-sheet type alignment of gelators with an interdigitated N-acyl unit. Gelation ability has also been affected by the conformational flexibility of the side chains, while the Tgel value decreases with the increase in hydrophobicity of the media. The increase in hydrophobic bulk of the αAA-side chain of the gelator increases the Tgel values in polar aprotic solvents. WAXRD studies reveal a Pm3m type cubic lattice being omnipresent in their xerogels without much alteration of the interdigitated molecular arrangement, even on such structural modification; however, d100 values decrease on going from apolar to aprotic polar media, whereas, it increases on going from aprotic polar to protic polar solvents (except 4c). Therefore, a substantial hydrophobic influence is presumably operative in aprotic polar media, but a competitive H-bonding interaction from polar protic solvent reduces the compactness of the network. A comparable influence of hydrophobicity is found for 4c and 4d in the xerogel state. Gelation from the achiral variety (4f), though weak, emphasizes that proper alignment of supramolecularly interactive moieties and solvophobic association play primary roles towards gelation, which is reinforced by chirality. The solvent effect on such systems was further analyzed using various solvent parameters and found to have an important role in such a self-assembly process.
Introduction
Supramolecular gels comprised of tunable functionalities have attracted much interest in a range of areas, including healthcare, environmental protection and energy-related technologies.1 However, they have become a challenging target in the non-covalent synthesis of nano-structural functional materials, towards the formation of uniform features that exhibit well defined properties with precise control over shape, size and stability.2 While surfactant self-assembly is primarily favoured by van der Waals stabilization of hydrophobic tails and the solvation of polar head groups, including electrostatic repulsions among similarly charged units, facilitating the curvature formation, and is also predicted by the polar–apolar ratio (the packing parameter) of their backbone architecture,3 the rationale behind gelation self-assembly, its stability and regulating ability is still, for the most part, attributed to serendipity and thus continues the quest for a systematic understanding of the logic behind such phenomena.4 Understanding the role of amino acid side chains helps in elucidating how tertiary structure depends on the primary sequence in a protein, where the mutations have been quantified in terms of the changes in stability they induce.5 Moreover, the state of aggregation or assembly of proteins imposes a serious impact on their function.6 Learning from nature, amino acids with diverse side chains have been widely exploited as synthetic scaffolds in supramolecular biomimetic systems, such as in peptidomimetics, peptide amphiphiles, amphiphilic peptides and also in miscellaneous applications as biocompatible and environmental friendly functional systems of nano-scale dimension,7 designing organized smart materials,8 controlled drug delivery systems,9 scaffolds for tissue engineering10 and also in the area of organocatalysis.11 Like polypeptides and proteins, such molecular self-assemblies ubiquitously involve various weak intermolecular interactions, such as hydrogen bonding, van der Waals interactions, π–π stacking interactions, dipole–dipole interactions12 and even metal–ligand coordination interactions.13 Owing to the presence of inherent chirality, with varied side chain residues associated with non-covalently interactive intervening amide linkages, the supramolecularity of amino acid derived systems give rise to anisotropic aggregation that provides amyloid-like fibrillar structures,14 which on subsequent physical cross linking among such fibrils, lead to the formation of supramolecular fibrous networks that restrict the flow of the entrapped solvent, resulting in gelation. Supramolecular gels are usually mechanically weak and unable to support the formation of free-standing structures for which their practical use with applied loads has been limited;15 to address this issue, a supramolecular gel from calixarene-derived networks has been very recently developed by Lee et al.16 Modulation of organogel properties has also been addressed by controlling the organic subphase composition,17 using mono- and bis-triterpenoids18 or cholesterol,19 or by isosteric substitution,20 producing different microstructures of assembly of the organogel derived systems. The primary driving force behind the self-assembly process of Fmoc-dipeptides constituted of phenylalanine residues is found to be the aromatic π–π interactions of the fluorenyl moieties, whilst hydrogen bonding has a secondary role, as reported by Tang et al.21 The molecular geometry of amino acid side chains, hydrophobic length and swapping between leucine and isoleucine are crucial for the development of their nanostructures, as reported by Han et al.22 A simulation study has been performed by Marchut et al. to elucidate the role of side chain interaction, which suggests the spontaneous formation of amyloid aggregates and annular structures from β-sheet type aggregates, rather than random configuration or coils.23 However, Dobson suggested that in amyloid fibrils the main chain dominates the structure, while the side chains are incorporated in the most favorable manner consistent with the requirements.24 Hydrophobic side chain residues have been found to provide much of thermodynamic driving force of the folding, self-assembly and consequent hydrogelation of amphiphilic β-hairpin peptides, as shown very recently by Micklitsch et al.25 The role of an adjacently placed phenylalanine unit in a pentapeptide based gelator on their gelation has been addressed by Banerjee et al.26 Dipeptides with aromatic protecting groups, such as Fmoc, have been found to have good gelation behaviour via H-bonding and π–π interaction.27 The presence of chirality is also an important parameter for directional anisotropic interaction for gelation, as described by Aggeli et al.28 Considering the entropy change associated with the hydrophobic effect, Escuder, Miravet and co-workers studied a family of bola-amphiphilic gelators with varied methylene spacers and found these to be the driving force for aggregation.29 The importance of the hydroxyl group of serine has been highlighted by Duarte et al.30 The role of side chain residues of amino acids in a micellar self-assembly has been addressed earlier by Miyagishi31 and our group.32 Towards the tuning of gelation properties, variable gelation time and stiffness of low-molecular-weight hydrogels was achieved through catalytic control over self-assembly by Poolman et al.33 Recently, the mechanical and morphological properties of self-assembled peptide hydrogels was attained via control over the gelation mechanism, through the regulation of ionic strength and the rate of pH change, by Li et al.34 A recent review, Tuning of Soft Nanostructures in Self-assembled Supramolecular Gels, From Morphology Control to Morphology-Dependent Functions, was done by Zhang et al.35
In connection to the contemporary interest of finding finer structural factors controlling the gelation ability, a unique gelator architecture capable of accommodating the essential modulation, yet exhibiting gelation self-assembly is required, which happens to be the bottleneck of the research in this direction. Despite the prevalence of serendipity36 in the design of gelator architecture, in our earlier work,37 it was successfully shown that the rational design of an amino acid based gelator with four distinct supramolecularly interactive segments can help in producing a gel network, of which the van der Waals interactions were modulated with different aliphatic spacers and N-acyl chain lengths. Nevertheless, the important aspect of finer modulation at the amino acid segment was not addressed in an elucidative fashion, as its understanding is not so straight forward and needs special attention with proper choice of amino acids. The main objective of the present paper deals with the rationalization of finer control of gelation properties through modulation of a single α-amino acid side chain in a dipeptide based organogelator in diverse media, both polar and apolar. With chirality being an important factor for amino acid based gelators, the issue of chiral influence on gelation is addressed, as well using an achiral variety. However, the gelation self-assembly in the present case can only emphasize the findings related to the effect of side chains in a peptidic motif with a single amino acid placed in a parallel β-sheet type arrangement and not the antiparallel one. Nevertheless, studies on the role of side chain functionalities in peptide structure–function properties have been previously attempted on various occasions with a series of hydrophobic amino acids38 in peptide based backbones in proteins; to our knowledge there has so far been no report of any kind that deals with their individual contribution, based on their inherent architecture, towards peptide based gelators. To address the issue, herein, the literature information related to side chains in protein chemistry has been considered and analyzed with the obtained data in various physicochemical studies and not by any instinctive understanding; however, it looks very simple and is not surprising to look at. Mother Nature took millions of years to optimize the molecular architecture of 20 natural amino acids, and therefore, very careful understanding is needed before commenting on their effect in an intuitive manner. Moreover, if an intuitive steric reason is considered to be the sole and trivial factor then it would be easier to design peptide based gelators with predictable properties, which is not so in reality. The manuscript thus highlights a plausible approach towards the fine tuning of gelation properties achievable via the simple modulation of an intrinsic structural element like changing a single amino acid's side chain encrypted on the gelator backbone architecture with its associated elements. With solvents being a major component in gel-systems, their influence on gelation is also discussed.
Experimental methods
Materials
Dicyclohexyl carbodiimide (DCC), α-amino acids (L-isomers of alanine, valine, leucine, isoleucine, phenylalanine and glycine) and β-alanine were purchased from Sigma Chemicals. n-Hexadecanoic acid and 1-hydroxybenzotriazole (HOBT) were purchased from SRL, India. N-Hydroxysuccinimide (NHS) and K2CO3 were purchased from Spectrochem, India. Analytical grade solvents were purchased from SD Fine Chemicals, India and were freshly distilled before use. Silica gel G was used for TLC and iodine vapour was used as a staining agent.
Synthesis
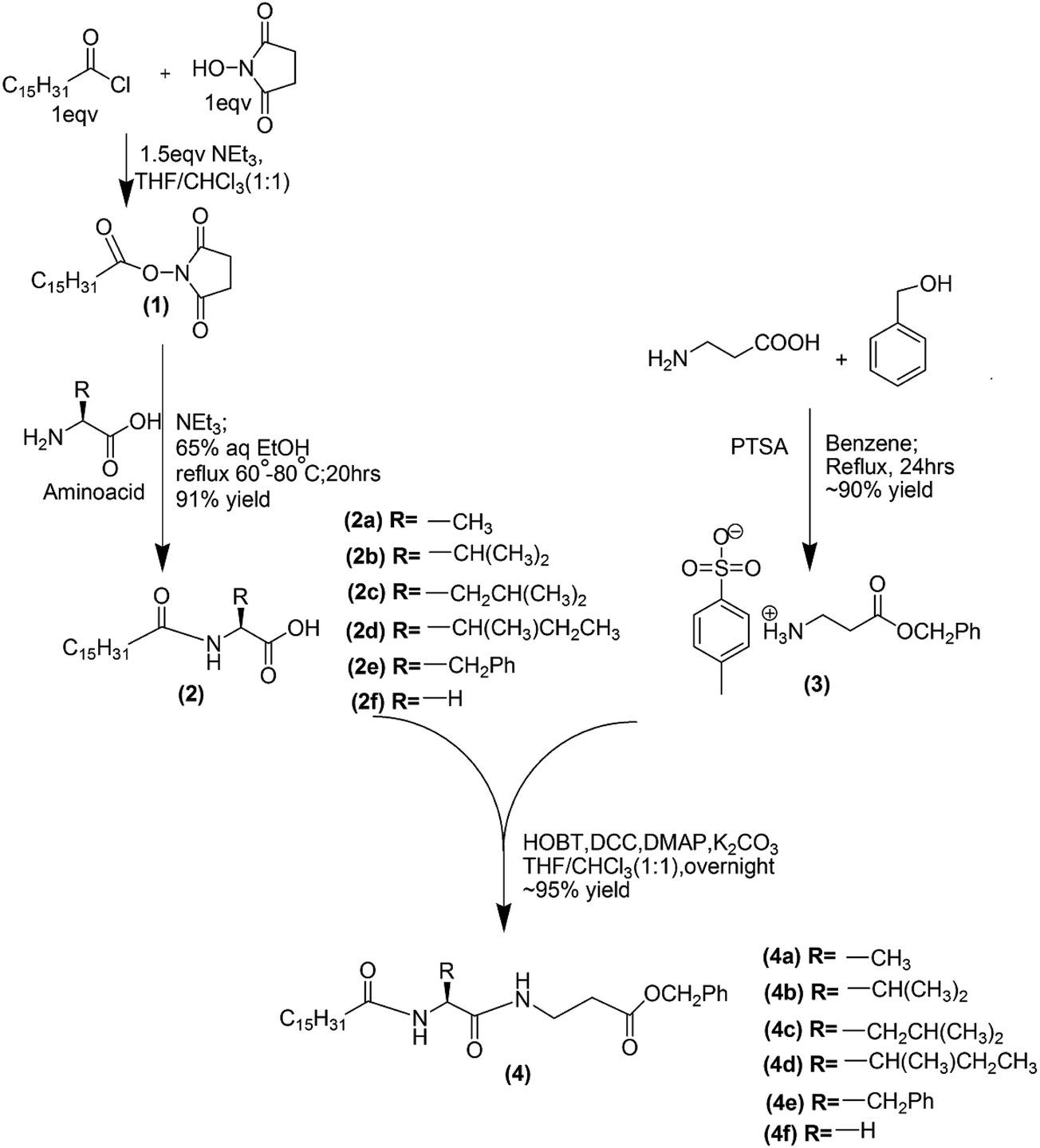
The dipeptide based gelators (4a–4f) have been synthesized in our laboratory following the given scheme (Scheme 1); the details of the synthetic procedures, 1H and 13C NMR, IR and mass spectroscopic data have been given in the ESI.†
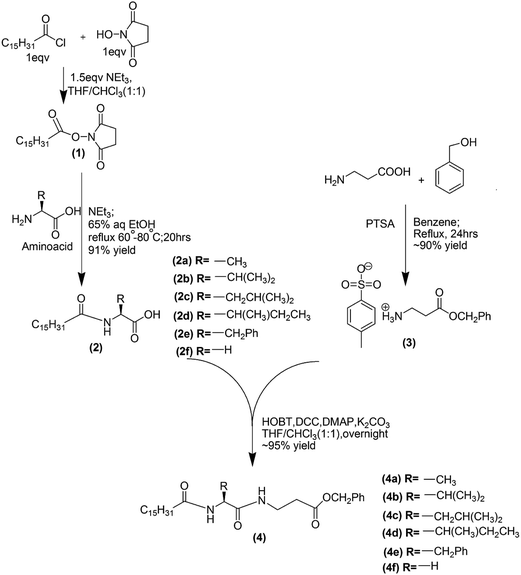 |
| | Scheme 1 | |
Determination of the critical concentration for gelation, by gelation number (GN)
The gelators used for the present investigation are usually insoluble in the majority of solvents at room temperature; however, they dissolved on heating and turned into gels on cooling to room temperature. The insolubility might be due to the extensive H-bonding across the amide units in their solid state molecular organization, which needed to be separated at higher temperature with solvation. The critical concentrations for gelation and subsequent gelation numbers (GN) were determined by dissolving a known amount of gelator in a specific volume of a solvent; gels were prepared in identical vials at room temperature and the amount of solvent required for gelation by the specific gelator was measured.
Determination of gel melting temperature
Gel to sol transition temperature (Tgel) was determined using the vial inversion method,39 as well as by oscillatory temperature-sweep rheology.40 In the former method, the Tgel was defined as the temperature at which the gel melts and begins to flow out, while for the latter method, the Tgel was taken as the temperature at which the elastic modulus (G′) drops below the viscous modulus (G′′), indicating the point at which the gel breaks under an applied stress. Tgel of the organogels was determined for various solvents at different concentrations and the obtained data corroborated each other considerably in either of the methods. The vials containing the gels were placed on a water bath with a high-precision temperature regulator and heated. The temperature at which the gel started to flow was taken as Tgel of the organogels. The bath was warmed at the rate of about 0.1 °C per s. In the rheological method, the gel melting temperature was measured by dynamic temperature sweep experiment, conducted for the gels in acetonitrile at CGC at strain amplitude of 0.01% and a constant frequency of 1 Hz. The test temperature was increased from 20 °C to 80 °C, at 2 °C per minute.
FT-IR spectroscopy
FT-IR studies were performed in a BRUKER ALPHA FT-IR spectrometer using the compounds in solution state in CHCl3 and in their gel state in acetonitrile.
WAXRD study of thin films of xerogels
Thin films of air dried gels (xerogels) were prepared by spreading soft organogel in acetonitrile on glass cover slips and drying carefully. The obtained films were subjected to wide angle powdered XRD studies using a Bruker D8 Advance instrument with a parallel beam optics attachment. The instrument was operated at 35 kV voltage and 30 mA current, using Ni-filtered Cu Kα radiation and was calibrated with a standard silicon sample. Samples were scanned from 2θ of 0° to 30° in the step scan mode (step size 0.020, preset time 2 s) and the diffraction patterns were recorded using a scintillation scan detector.
UV-Vis and fluorescence
UV-Vis and fluorescence spectroscopic studies were performed with a SpectraMax M5 spectrophotometer, using 1 cm quartz cells.
Circular dichroism
CD spectra were recorded on a Jasco (model J-815) CD spectrometer with a band width of 1 nm and slit width of 100 μm in the ultraviolet region (200–350 nm), taking the sample in a quartz cuvette of 1 mm path length. Experiments were performed with an interval of 0.5 nm, with a scan speed of 200 nm per min at a controlled temperature of 20 °C. The samples for the experiment were prepared at different concentrations as required.
Field emission scanning electron microscopic study
Morphologies of the gel materials were investigated using a FESEM instrument, INSPECT F50 (Netherland) with an acceleration voltage of 20 kV. For this study, samples of the xerogels were prepared on a glass slide, scratched carefully, placed on carbon tape and coated with a thin layer of gold.
Rheology
Frequency sweep rheological experiments were performed at 25 °C using an MCR-102 rheometer (Anton Paar) with a cone-plate (40 mm diameter and cone angle 1 degree) attachment at a gap setting of d = 0.080 mm, linear frequency range of 1.0 to 10.0 Hz and constant strain of 0.01%. Rheological oscillatory strain sweep experiments were also executed with the same instrument at a strain amplitude (γ) ranging from 0.01% to 100% and a constant angular frequency of 10 rad s−1.
Results and discussion
Design of gelator molecules
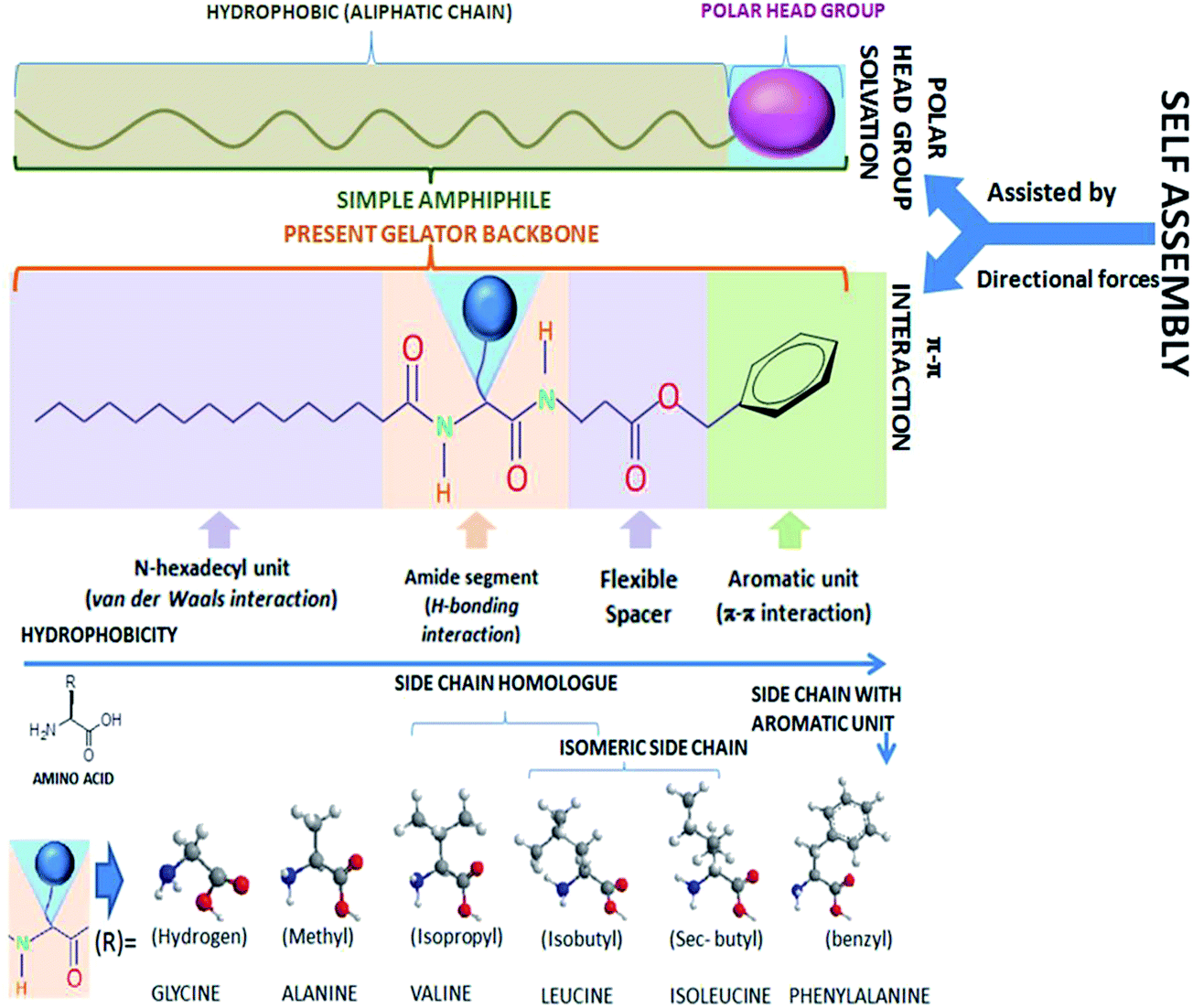
Due to intrinsic structural and functional intricacies, the role of naturally occurring amino acids with distinct side-chains embedded in a peptide based gelating system remains critical. To address such an issue, a gelator backbone architecture is required, where an amino acid can be changed and yet maintain its gelation ability; the lack of such has limited these studies in the literature. Herein, the design is based on principles of amphiphilic self-assembly, where the polar unit of the amphiphilic backbone has been replaced with an aromatic functionality, with a vision that the primary alignment of the molecule might occur through intergelators; the π–π type directive interaction in a particular orientation in organic media, which is similar to the solvated polar head groups, protrude in one direction in aqueous dispersions of amphiphiles (Fig. 1). The structure of the gelator consists of four distinct functional zones: (i) an aromatic unit (benzyl ester) placed at one end of the molecular backbone, (ii) a dipeptidic unit arising from peptide bonds between α-amino acid (α-AA) and (β)-alanine residues, (iii) a flexible hydrophobic ethylene spacer sandwiched between aromatic and peptidic zones and (iv) includes a n-hexadecanoyl chain connected to the N-termini of the dipeptide (Fig. 1). These four segments exhibit specific supramolecular interactions based on their inherent functionalities. A π–π type interaction is apparently evident at segment (i), and H-bonding interaction is expected from segment (ii). However, a van der Waals type interaction is likely to be predominant at the segments (iii) and (iv). A set of six naturally occurring hydrophobic α-amino acids (α-AA) have been chosen for the present study and were embedded in the molecular backbone, giving rise to six gelators: 4a–4f with a common general formula, N-n-hexadecanoyl-αAA-(β)Ala-OBn [αAA = L-isomers of Ala (4a), Val (4b), Leu (4c), Ile (4d), Phe (4e) and Gly (4f)]. The chosen hydrophobic amino acids (α-AA) differ on a hydrophobic scale, as depicted in the literature by Jones and Middelberg,41 representing the following trend: Gly (0.501) [4f] < Ala (0.616) [4a] < Val (0.825) [4b] < Leu (0.943) [4c] ≈ Ile (0.943) [4d] < Phe (1.00) [4e], comprised of side chains of hydrogen atoms, methyl, isopropyl, isobutyl, sec-butyl and benzyl groups, respectively (Fig. 1). In addition to the hydrophobicity scale, the geometry of the side chains is also noteworthy. The accessible surface area (ASA) in Å2 of such residues based on glycine are found to be 67 (Ala), 117 (Val), 137 (Leu) 140 (Ile) and 170 (Phe). The steric bulk of the Cα-substituents are correlated to the Voronoi volumes of the residues in Vm in cm3 mol−1, which are as follows: Ala (15.4 ± 2.1) < Val (44.8 ± 2.9) < Leu (59.8 ± 3.5) ≈ Ile (59.7 ± 3.2) < Phe (76.5 ± 2.9), while Gly has a substantially higher Voronoi volume of 38.4 ± 1.6. Such a higher value for Gly is attributed to the combined space of van der Waals volume of the residue, plus the void space around it, where the latter reflects the packing defect.42 The substitution pattern at the side chains are also of significance: 4c with leucine residue consists of a side chain (isobutyl) that is just a higher homologue of the side chain of valine (isopropyl), which is present in 4b as its αAA, whereas, 4c and 4d possess constitutionally isomeric side chains, i.e., isobutyl and sec-butyl, respectively. A closer look at the side chains between 4b and 4d reveals that, the Cβ carbon possesses two methyl groups in the case of 4b, whereas 4d consists of one methyl and one ethyl group, as if the γ-carbon of 4b has an extra methyl substituent in 4d. On the contrary, 4e (with the phenylalanine unit) consists of a distinctly different side chain than the others, i.e., an aromatic side chain with a benzyl residue. 4e is also unique because the side chain can provide an inter-amino acid attractive type π–π interaction, whereas, in all other cases, it is the van der Waals repulsion that can significantly be operative. The choice of taking up the achiral variety of the gelator 4f with Gly as α-AA is based on the rationale that it can not only give the comparable effect of Cα substitution at side chains with respect to H-atom, but it can also address the importance of chirality on gelation, if any, pertaining to gelator architecture. Since the molecular arrangement in the self-assembly is envisaged to be the parallel β-sheet type, we have also taken into account the β-sheet propensity factor of Chao and Fasman of the respective amino acids: Gly (0.75), Ala (0.83), Val (1.70), Leu (1.30), Ile (1.60) and Phe (1.38). Despite Leu and Ile possessing isomeric side chains, the β-sheet propensity factor is comparatively higher in the case of Ile than Leu. Such differences might originate from the side chain flexibilities and to rationalize that we got the help of the conformational flexibility scale (based on short structureless peptides), suggesting the decreasing order of flexibility is Gly > Ala > Leu > Phe > Val > Ile43 (as proposed by Huang et al.), and also the partial molar isentropic pressure coefficient values (in 10−4 cm3 mol−1 bar−1) of the side chain residues (with respect to glycine) as depicted earlier: Phe (5.8) > Ile (4.3) > Ala (3.7) > Val (1.8) > Leu (1.3), which indicates that between isomeric Leu and Ile, a larger value for the isoleucyl side-chain results from a high degree of hydrophobic influence towards disruption of the peptide group hydration in aqueous media. The loss of entropy from the conformational changes plays a significant role in a self-assembly process. Thus, the conformational changes concomitant with the amino acids embedded in a peptide based gelator backbone can be envisaged to play an important role as well.
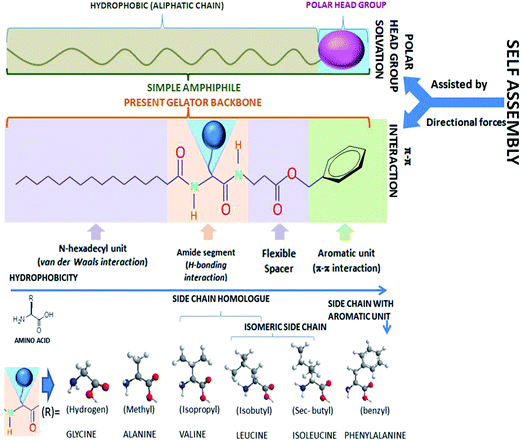 |
| | Fig. 1 Schematic of the gelators' backbone architecture. | |
Morphology
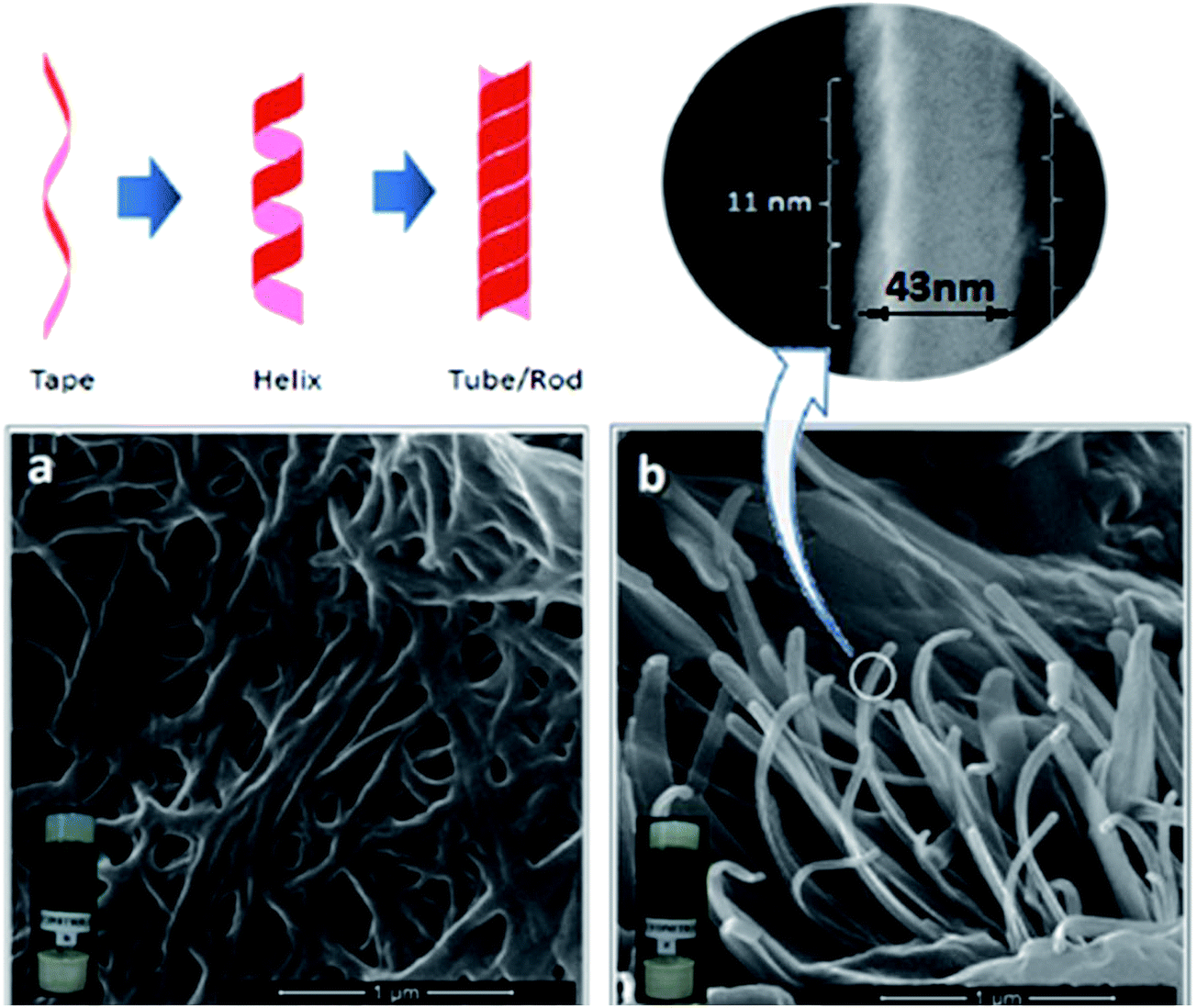
Field emission scanning electron microscopy (FESEM) was employed to discern the self-assembled 3D networks existing in the gel matrices, and all of them showed fibrous nanostructured networks, of which two representative images of 4b and 4e are given (Fig. 2). The formation of such organized nanostructures has been widely found in supramolecularly interactive organogels.44,45 More complex fibrillar structures, such as double tapes and fibrils in different amyloid systems, have been shown to possess helicoidal, cylindrical helical and tubular geometries.46 A diverse range of helical gels based on chiral self-assembling elements, such as chiral H-bond donor/acceptor groups like urea47 and amide,48 have been accounted for. In the present study, a similar observation was also explicated in either of the cases (4b and 4e); a fibrillar network with a rod like architecture was found to possess helical fibres of an average pitch length of 11 ± 2 nm, with a width of 43 ± 5 nm, as observed in higher resolution; however, the morphologies are not found to be significantly different, even in the presence of differences in backbone architecture (Fig. 2).
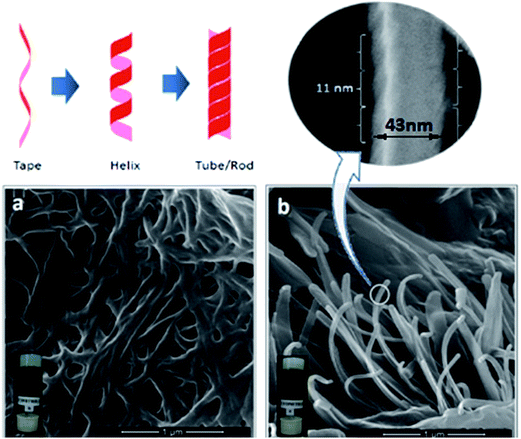 |
| | Fig. 2 FESEM images of organogels (a) 4b and (b) 4e in acetonitrile at the xerogel state (inset represents the digital images of respective organogels). | |
Gelation behaviour
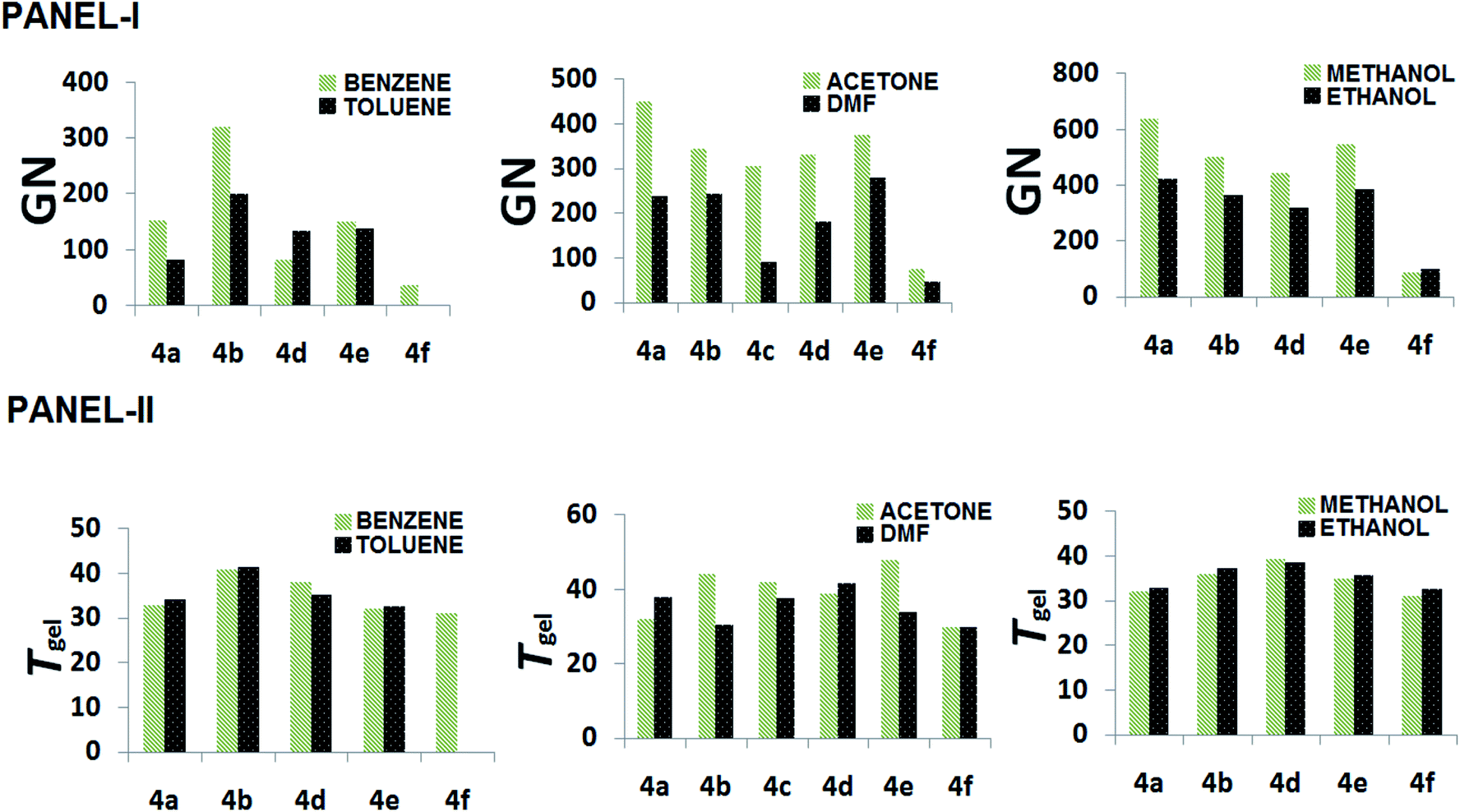
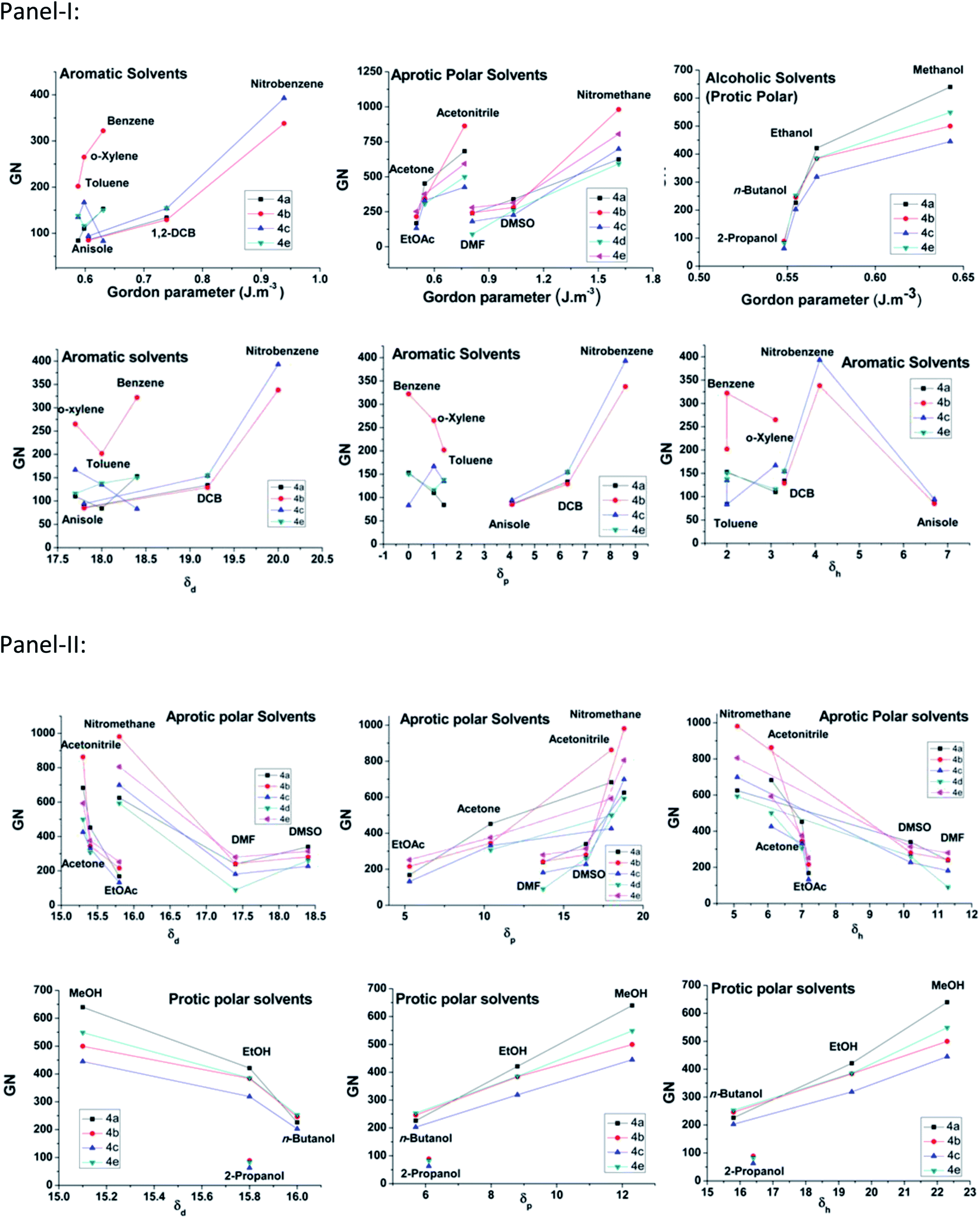
The gelators have shown ambidextrous gelation ability in various polar and apolar solvents, including aqueous alcoholic solutions (Table 1). The gels are stable for weeks and do not transform into viscoelastic fluids or crystallize on prolonged storage in stoppered vessels at room temperature. The critical gelation concentration (CGC) has been expressed in terms of gelation number (GN),49 which is defined as the number of solvent molecules entrapped in the gel matrix per gelator molecule. The GN values have been found to vary in different solvents, as well as across the gelator architecture. In general, gelators show significantly higher gelation ability in polar media, both protic and aprotic, such as EtOAc, DMF, acetonitrile, DMSO, n-butanol, and an exceptionally high GN in nitromethane, which is in concurrence with the polarophobic interaction that might be a dominant factor behind their overall gelation. Keeping our primary attention towards the effect of the amino acids' Cα-substituents, it was observed that 4f, with an achiral backbone with glycine amino acid, shows the weakest gelation ability amongst all. While comparing the gelation ability among the others, it was observed that 4c (iso-butyl) showed gelation ability in a limited number of solvents, whereas, 4b (isopropyl) and 4d (sec-butyl) exhibited gelation in a wide range of solvents. The usual trend of GN in three different types of solvents, aromatic, aprotic polar and protic polar, is presented in Fig. 3 (panel-I). Considering the hydrophobicity as a primary criterion, the decreasing trend of GN in polar solvents (protic or aprotic) is supposed to be 4e > 4d > 4c > 4b > 4a > 4f. However, the obtained results do not follow the expected trend, i.e., the trend in EtOAc is 4e > 4b > 4a > 4d > 4c > 4f and in CH3CN it is 4b > 4a > 4e > 4c > 4d > 4f, whereas, in DMF 4e > 4b ≈ 4a > 4d > 4c > 4f; 4a and 4b show much better gelation than expected. Thus, hydrophobicity cannot be the sole reason for the differences in gelation ability among the gelators. This could be due to the difference in molecular packing arising from optimal side chain volume and its inherent conformational flexibility. The high gelation ability of 4e is attributed to the intergelator π–π type network of the side chains, along with its hydrophobicity; on the contrary, for 4f, the loss of chirality, as well as side chain hydrophobicity, limit its gelation behaviour. The solvent–gelator π–π interaction is presumably more effective with the electron acceptor solvents like nitrobenzene for the electrostatic effect, whereas, such interaction is repulsive with electron donor solvents like benzene, toluene or anisole.50 Thus, an electron deficient aromatic nucleus like nitrobenzene can destabilize the extensive π–π network of 4e, resulting in the loss of gelation. Nevertheless, the mutual assistance of hydrophobicity and H-bonding interaction might overcome the solvation effect for the gelation of 4b and 4d, the polarophobic effect with the optimum molecular packing presumably fails to gelate 4a and 4c in nitrobenzene. To rationalize the results, we took the analogy of a methodical approach of Bromberg et al. towards finding the effect of the packing of amino acid side chains on protein stability and structure, exploiting the lattice model with side chains (SCM) and a linear lattice model without side chains (LCM). According to them, the comparison shows that (1) side chain degrees of freedom increase the entropy of open conformations, but side-chain steric exclusion decreases the entropy of compact conformations, thus producing a substantial entropy that opposes folding; (2) there is a side-chain “freezing” or ordering, i.e., a sharp decrease in entropy near maximum compactness; (3) the different types of contacts among side chains(s) and main-chain elements (m) have different frequencies, which have different dependences on compactness. The main chain–main chain contacts (mm) contribute significantly only at high densities, suggesting that main chain hydrogen bonding in proteins may be promoted by compactness.51 Correlating such inferences with the present study, the obtained results exhibit a significant corroboration. The Voronoi volumes (Vm in cm3 mol−1) have been found to increase with the bulk of the side chains, e.g., Ala (15.4 ± 2.1) < Val (44.8 ± 2.9) < Leu (59.8 ± 3.5) ≈ Ile (59.7 ± 3.2) < Phe (76.5 ± 2.9), while Gly has a substantial higher Voronoi volume of 38.4 ± 1.6. Such a high value is attributed to the combined space of van der Waals volume of the residue, plus the void space around it, where the latter reflects the packing defect. To avoid such discrepancies, the van der Waals volumes of the side chains of the amino acids were also compared. Based on the side chain volume (in Å3) of glycine, the side chain volumes are as follows: 26.3 (Ala) < 75.3 (Val) < 100.8 (Leu) < 101.1 (Ile) and 129.7 (Phe), as obtained from the work of Harpaz et al.52 Thus, the steric repulsions are expected to follow the similar trend during molecular packing of their self-assembly.42 Between the two extremes of the jigsaw puzzle model (specific) and the ‘nuts and bolts’ model (non-specific)51 of the packing of amino acids in proteins, we can envisage that the former packing mode is apt for the present system of homomeric self-assembly, comprising of a single amino acid based parallel beta-sheet type arrangement of gelators. Thus, based on the fact that better packing leads to better stability for proteins53 with polymeric peptidic networks, such dependence can also be verified to validate the self-assembly of the n-meric peptidic network of the gelators. The obtained results significantly corroborate the stability of gels based on the packing of the gelators, which differs only at the variation of the side-chains. In general, 4a, with the methyl group at the side chain providing the least steric repulsion, shows better gelation self-assembly, having higher GN values. However, steric interaction was not the sole criterion determining the efficacy of the gelation process. The increase in steric bulk not only increases the side chain-side chain repulsion, but also, the conformational flexibility pertinent to the side chain spatial geometry influences the packing and eventually affects the gelation self-assembly that reduces the GN values. The effect of such flexibility is clearly demonstrated for gelators with homologous side chains, 4b and 4c, where the gelation ability is weaker for the higher homologue 4c. Although the increase in the side chain hydrophobic surface for 4c helps in gelation in polar aprotic media, the presence of an extra methylene spacer in 4c gives rise to higher conformational flexibility than in the case of 4b, which seemingly hinders the molecular packing, resulting in lower gelation ability for 4c compared to 4b. A remarkable difference in gelation behaviour has been witnessed for gelators with isomeric side chain residues in 4c and 4d. A significantly weak gelation ability was found for 4c, which shows gelation in very few solvents under investigation. The displacement of one of the methyl groups from Cγ of 4c to Cβ results in the 4d architecture, where the results show that such modification significantly reduces the gelation ability. Therefore, anything bigger than the isopropyl substituent (with β,β-dimethyl substitution) hinders the gelation ability in a drastic fashion, although flexibility might compensate to a smaller extent, but not as good as an isopropyl unit (as in 4b). The analysis of the obtained data also accounts for the unique molecular arrangement with a peptidic motif having a single amino acid placed in a parallel β-sheet type orientation. Comparing the GN values in nitromethane (aprotic polar) and 80% EtOH–H2O, it can be visualized that the hydrophobic complementarity of the side chains follows the trend 4b > 4e > 4d > 4c > 4a > 4f in the former, whereas in protic polar media, a suspected interaction of solvents in H-bonding amide segments, the overall hydrophobic collapse would lead to better packing, even with lower bulky substituents and the trend becomes 4a > 4b > 4d > 4e ≈ 4c > 4f in 80% EtOH–H2O.
Table 1 Gelation number and gel to sol phase transition temperature (Tgel) of gelators in different solvents
| Solvents |
4a |
4bb |
4c |
4d |
4e |
4f |
| GN |
Tgel |
GN |
Tgel |
GN |
Tgel |
GN |
Tgel |
GN |
Tgel |
GN |
Tgel |
| DCB = O-dichlorobenzene, PE = petroleum ether, EG = ethylene glycol, s = soluble, is = insoluble, wg = weak gel. Ref. 49. |
| Benzene |
153 |
33 |
322 |
40.9 |
s |
s |
83 |
38.2 |
151 |
32.2 |
38 |
31.2 |
| O-Xylene |
110 |
34 |
265 |
41.3 |
112 |
33.4 |
167 |
39.5 |
116 |
32.5 |
s |
s |
| Toluene |
84 |
34.2 |
202 |
41.4 |
s |
s |
135 |
35.3 |
138 |
32.7 |
s |
s |
| DCBa |
134 |
40.6 |
129 |
44.2 |
s |
s |
154 |
36.2 |
155 |
37.2 |
75 |
30.2 |
| Anisole |
86 |
41.3 |
85 |
39.1 |
s |
s |
94 |
41.8 |
s |
s |
s |
s |
| Nitrobenzene |
s |
s |
338 |
40.1 |
s |
s |
393 |
42.1 |
s |
s |
s |
s |
| EtOAc |
168 |
36.8 |
216 |
38.0 |
134 |
35 |
132 |
43.1 |
253 |
32.6 |
53 |
30.6 |
| DMF |
239 |
38 |
243 |
30.5 |
90 |
37.7 |
181 |
41.7 |
280 |
34.0 |
48 |
31.0 |
| DMSO |
340 |
61.3 |
281 |
54.3 |
261 |
64.5 |
227 |
63.7 |
314 |
43.1 |
58 |
31.1 |
| Acetonitrile |
683 |
51.1 |
863 |
47.9 |
500 |
64.0 |
426 |
62.0 |
593 |
42.0 |
87 |
32.0 |
| EtOH |
421 |
33 |
364 |
37.2 |
wg |
wg |
319 |
38.7 |
385 |
35.7 |
98 |
32.7 |
| Acetone |
452 |
32 |
345 |
44.2 |
307 |
42.0 |
331 |
38.9 |
376 |
48.0 |
76 |
30.0 |
| MeOH |
640 |
32.1 |
500 |
36.0 |
wg |
wg |
445 |
39.3 |
549 |
35.0 |
86 |
31.0 |
| Nitromethane |
625 |
48.5 |
981 |
49.8 |
593 |
49.9 |
699 |
51.8 |
806 |
44.5 |
80 |
32.5 |
| PE |
s |
s |
64 |
58.8 |
s |
s |
58 |
56.3 |
54 |
57.0 |
s |
s |
| THF |
s |
s |
s |
s |
s |
s |
s |
s |
s |
s |
s |
s |
| Dioxane |
s |
s |
s |
s |
s |
s |
s |
s |
s |
s |
s |
s |
| 50% aq. EG |
s |
s |
s |
s |
s |
s |
s |
s |
s |
s |
s |
s |
| Chloroform |
s |
s |
s |
s |
s |
s |
s |
s |
s |
s |
s |
s |
| 50% aq. glycerol |
s |
s |
s |
s |
s |
s |
s |
s |
s |
s |
is |
is |
| Isopropanol |
s |
s |
89 |
32.5 |
s |
s |
63 |
31.7 |
83 |
32.3 |
s |
s |
| Butanol |
226 |
33.6 |
246 |
34.8 |
s |
s |
203 |
33.2 |
253 |
33.1 |
63 |
30.3 |
EtOH/water (80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20) :20) |
563 |
35.9 |
491 |
35.6 |
468 |
35.2 |
479 |
34.7 |
448 |
34.4 |
105 |
31.4 |
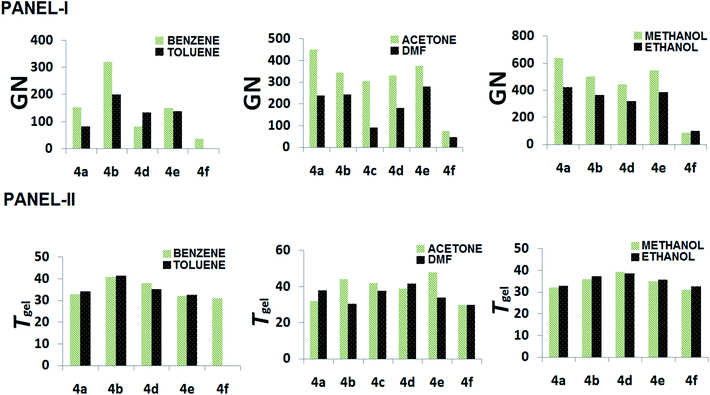 |
| | Fig. 3 Panel-I represents the plot of the GN of different gelators in various solvents (aromatic, polar aprotic and protic) and panel-II represents the plot of Tgel of different gelators in different solvent systems. | |
Thermal stability of gels
In general, thermal stability means the stability at higher temperature. Gel to sol transition is a cooperative effect of different non-covalent interactions; ideally, to understand the role of only one parameter (e.g. the effect of side chains), all the other parameters need to be kept identical. Since a small change in molecular architecture can affect the physicochemical properties in a remarkable fashion, like H2O and D2O,54 in a realistic approach, we tried to deal with the issue of side chain modulation in a carefully designed gelator backbone with a single variable in the molecular structure (i.e., side chains of six hydrophobic α-amino acids) and analyzed their thermal stability in a solvent specific manner. On analyzing the obtained data, we can see that (i) 4f shows the lowest Tgel values, irrespective of solvent; (ii) in aromatic solvents like benzene, toluene and o-xylene, 4e shows much lower Tgel values than in nitrobenzene or o-dichlorobenzene; (iii) in aprotic polar solvents like DMSO, DMF, CH3CN and nitromethane, the decreasing trend of gel stability is apparently 4d > 4a > 4c > 4b (DMF), 4c > 4d > 4a > 4b (DMSO), 4c > 4d > 4a > 4b (CH3CN); 4d > 4c > 4b > 4a > 4f (nitromethane); (iv) (a) in protic polar solvents, Tgel decreases with the increase in solvent hydrophobicity, i.e., Tgel in methanol > ethanol > butanol; (b) for any particular alcoholic media, 4d > 4b > 4a (MeOH); 4d > 4b > 4a (ethanol) and 4b > 4a ≈ 4d (butanol), and finally, (c) in 80% (v/v) aq. ethanol, Tgel values are very close, 4a ≈ 4b ≈ 4c > 4d ≈ 4e. The lower value for 4f significantly suggests a strong influence of chirality that enhances the thermodynamic stability of the gelation self-assembly. 4e with the amino acid having aromatic side chains, gets very much affected with the aromatic solvents that apparently interact at the side chain-side chain π–π interaction and destabilizes the network and loses the gelation ability in anisole or nitrobenzene. In aprotic polar solvents, the hydrophobic effect plays an important role as 4c and 4d show much higher values of Tgel compared to 4b, whereas 4a shows more interesting behaviour with lower hydrophobic side chains. The most interesting results were obtained in nitromethane, where the decreasing trend of Tgel corroborates exactly with thy hydrophobicity of the amino acid side chains. In polar protic solvents, we do see three different kinds of effects. First of all, the Tgel value decreases, irrespective of the gelators, with the increase in hydrophobicity of the media, shown in Fig. 3 (panel-II). Secondly, the similar hydrophobic factor of the side chain plays a similar role, as found in aprotic polar solvents. The Tgel values in polar protic media are less than for the aprotic polar ones, which suggests that the amide functionality is under stress in protic polar solvents. Unique data are obtained in 80% (v/v) aq. ethanolic media, where the Tgel values are comparatively low and very similar for all the systems, which suggests that the major contribution comes from the aliphatic chain melting and the role of side chains is not significant. Since the physical gels are composed of non-covalent interactions of a different nature, the rationalization of thermal stability based on only one criterion is complex. In a β-sheet organization, the side chain residues are normally placed orthogonal to the backbone–backbone H-bonding plane. However, the stability is majorly influenced by the strength of the H-bonding, as well as inter-strand side chain-side chain interactions. The invariant nature of the H-bonding unit across the gelator architecture suggests that the difference in stability is attributed to the physicochemical differences arising from the geometry of the side chains. Both the steric bulk and the conformational flexibility of the side chains reflect the Tgel values, which might rationalize the small discrepancies in the trend as observed. The role of solvents at the molecular level is also to be taken into account, concentrating on their polar factors and is treated separately. While comparing the GN with the Tgel, it does not always go hand in hand. However, for a particular solvent, concentration dependent changes in Tgel values can give us an idea about the critical value of the growth of the fibrous network and its entanglement, which is reflected in the Tgel values.55 In a concentration dependent study, the gel to sol transition temperature (Tgel) was found to increase with the increase in concentration of gelators. For gel systems, both polymer gels and organogels, the concentration dependence of the gel–sol phase transition temperature has often been analyzed by means of the van't Hoff relationship.56 Therefore, it is very well known in the literature that concentration dependent changes in Tgel can account for their thermodynamic properties. Considering that the enthalpy of gelation remains the same, with a specific temperature range of the self-assembly process, n(gelator) + solvent ![[left over right harpoons]](https://www.rsc.org/images/entities/char_21cb.gif) gel.
gel.
The G° for the above process can be written as follows:
G° = −RTlnK = −RTln[gelator] (considering the activity of pure gelator and solvent to be unity).
If we consider that the enthalpy of the above process remains constant, then the van't Hoff enthalpy can be obtained from the following equation:
| H°(van't Hoff) ≈ −R{dlnK/d(1/T)} ≈ −R{dln[gelator]/d(1/T)} |
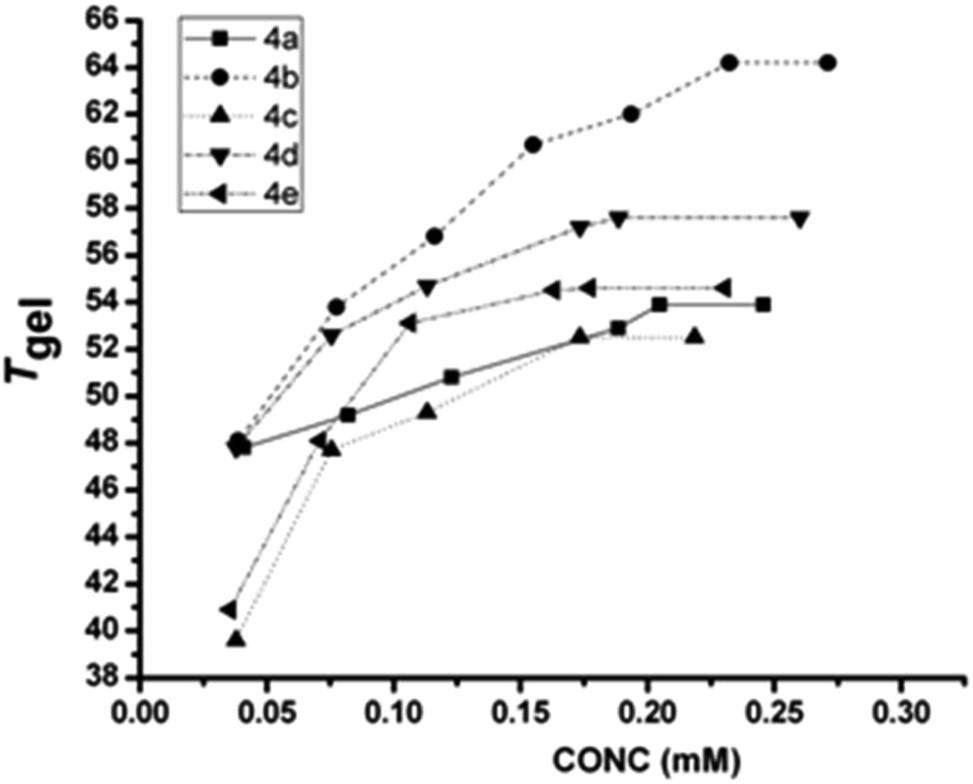
Therefore, from the slope of the plot, ln[gelator] vs. (1/T), the thermodynamic parameters (ΔH, ΔS and ΔG)57,58 were calculated at 298 K for gels from different gelators, a representative plot of which is given in Fig. 4 (extensive plots are given in the ESI, Fig. S1–S4†). Table 2 represents the results for three different solvents for the gelator 4a–4f. The results clearly show that in polar media (in acetonitrile and 80% (v/v) ethanol in water), the free energy decreases with the decrease in steric bulk of the side chains; the decrease in free energy is 4a > 4b > 4c > 4d and this trend is more pronounced in polar media (acetonitrile and 80% ethanol in water) than in apolar media like o-xylene. H-Bonding interactions among gelators are more decisive in apolar solvents, and hydrophobic interactions in protic polar media. Since acetonitrile is polar-aprotic, both H-bonding and the hydrophobic effect act simultaneously, which helps in the lowering of free energy and thus, better gelation is obtained, compared to that in o-xylene (apolar) and 80% ethanol in water (polar protic). 4e with a benzyl side chain shows the best favourable free energy for gelation, which is likely due to the additional π–π type attractive interaction arising from its side chins. In the case of 4f, the values of all three thermotropic parameters are in the lower range, suggesting its weaker self-assembly phenomena. However, the gel–sol transition temperature (Tgel) does not follow a similar trend as is observed for free energy values. The contribution of amino acid side chains to the thermodynamic parameters in polar protic solvents (EtOH and 80% aq. EtOH) can also be correlated to the partial molar isentropic pressure coefficient at infinite dilution, which is a quantity that is a sensitive indicator of the subtleties of solute–solvent interactions. The data by Hedwig suggest that in aqueous media, the contribution goes in the following order: Phe (5.8) > Ile (4.3) > Ala (3.7) > Val (1.8) > Leu (1.3) [in 10−4 cm3 mol−1 bar−1]. The free energy contribution based on glycine (4f), is Phe (12.1) (4e) > 11.4 (Ala, 4a) > 9.6 (Val, 4b) ≈ 9.4 (Leu, 4c) ≈ 9.3 (Ile, 4d). However, the differences among 4b, 4c, 4d is majorly pronounced in their enthalpic and entropic contributions. This could be due to the loss of entropy from the conformational changes in the self-assembly process, where the free energy of the residue changes for transferring a residue from the coil to the secondary structure in which all rotamers are populated;59 it is also known that the type of secondary structure of which a residue is part, generally has a significant impact on how much side-chain entropy is lost.60 We can see that the entropic contribution is very high in the case of 4c, compared to its isomeric 4d in either of the solvents (polar as well as nonpolar) and is attributed to the higher side chain flexibility associated with 4c. The obtained result is nicely corroborated by the study by Huang et al., where the side chain flexibility accounts for the kinetic measurements of the collision frequency between the two ends of short, random-coil polypeptides, which suggest the following order of flexibility: Gly > Ala > Leu > Phe > Val > Ile. Despite similar hydrophobicity, Voronoi volumes and surface areas between 4c and 4d, it is the side chain flexibility that contributes a lot to the thermodynamic factors, which also reflect on their mechanical strengths (discussed under rheology). In general, Tgel was found to be higher in aprotic polar media like DMF, acetonitrile, DMSO and nitromethane than in aromatic (benzene, toluene, o-xylene or anisole) and in protic polar media (methanol, ethanol, etc.). This suggests that the effect of the hydrophobicity of the substituents contributes significantly. Therefore, a sole factor is insufficient to rationalize all the experimental results; rather, a combined influence is important. A conformational reorganization across the gelator molecules in the gel network, as evident from CD studies (discussed later), might be an important reason behind such observation of Tgel data as in our earlier studies.37
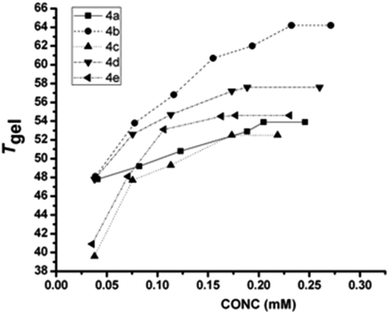 |
| | Fig. 4 Tgel (°C) versus concentration (mM) plot of different gelators in acetonitrile. | |
Table 2 Calculated thermodynamic parameters of the gelators in xylene, acetonitrile and (80:20) ethanol–water at 298 K
| |
4a |
4b |
4c |
4d |
4e |
4f |
| −ΔG° (kJ mol−1) |
Xylene |
12.3 |
10.2 |
9.4 |
10.4 |
17.7 |
— |
| CH3CN |
24.4 |
23.9 |
18.4 |
12.8 |
13.4 |
3.8 |
| EtOH/water (80:20) (v/v) |
16.3 |
14.5 |
14.3 |
14.2 |
17.0 |
4.9 |
| −ΔH° (kJ mol−1) |
Xylene |
87.4 |
115.2 |
136.1 |
130.7 |
142.3 |
— |
| CH3CN |
227.0 |
185.2 |
143.9 |
99.9 |
96.0 |
37.4 |
| EtOH/water (80:20) (v/v) |
93.0 |
71.1 |
85.5 |
70.2 |
80.2 |
49.8 |
| −ΔS° (J mol−1) |
Xylene |
252.0 |
342.0 |
425.0 |
400.0 |
419.0 |
— |
| CH3CN |
680.1 |
540.9 |
421.5 |
292.4 |
277.2 |
112.5 |
| EtOH/water (80:20) (v/v) |
259.0 |
190.0 |
239.0 |
188.0 |
212.0 |
150.1 |
Rheology
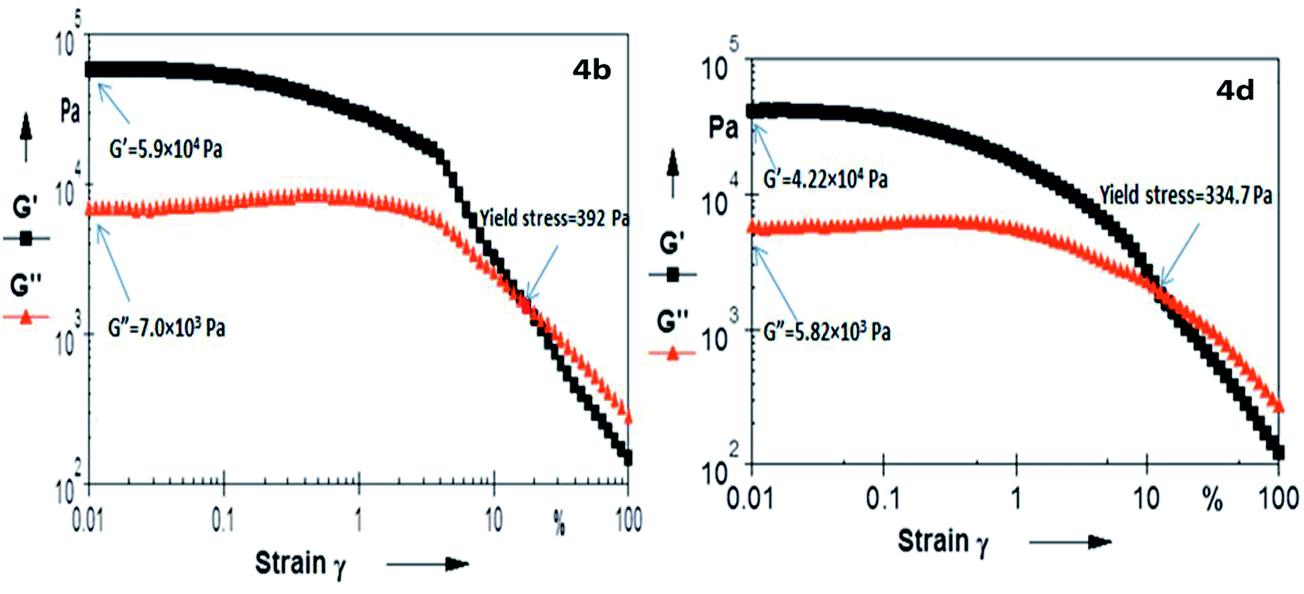
A quantitative estimation of the viscoelastic properties of the materials was carried out by oscillatory rheological experiments. From an experimental point of view, a gel is composed of two components (solvent and gelator), and is a viscoelastic system that exhibits two distinct mechanical properties: (i) the system should not flow under mechanical stress less than a limiting value, called yield stress and (ii) it must be more elastic than viscous.61 The viscoelastic response of the material is characterized by two material measurements: the elastic storage modulus G′(ω) and the viscous loss modulus G′′(ω), which are measured by dynamic oscillatory rheological strain sweep experiments.62 This study gives an indication towards the flow behaviour and rigidity of the gel. The storage modulus (G′) represents the ability of the deformed material to store energy, and the loss modulus (G′′) corresponds to the flow behaviour of the material under stress. These are the two main parameters for the evaluation of the mechanical strength of the gel system, where G′ > G′′ and G′′ > G′ at gel state and sol state, respectively. The crossover point between G′ and G′′ indicates the transition point from elastic solid to viscous liquid, while the yield stress is defined as the minimum stress to be applied to a sample in order to induce flow. This is determined by strain sweep experiments and the yield stress values (in Pa) for 4a–4f were found to be (4a) 459.5, (4b) 392, (4c) 356, (4d) 334, (4e) 916 and (4f) 122. A representative plot of the strain sweep experiment has been given in Fig. 5 (ESI, Fig. S5–S8†). The high yield stress value of 4e strongly suggests a better packing in their gelation self-assembly. Valine, with isopropyl side chain, can experience lower van der Waals repulsion, and its lesser flexibility helps for better packing, which results in the greater mechanical strength of the assembly, compared to others. Similarly, isoleucine with very high Voronoi volume and higher flexibility gives rise to the weaker mechanical strength of the gel network. Glycine, with the least steric repulsion from the side chain is expected to have the best packing; but the loss of the hydrophobic component and the larger void space in its Voronoi volume affect its packing. However, in the case of Phe, the higher flexibility and higher Voronoi volume is expected to have lower yield stress of the gel network. The opposite effect is ostensibly coming from the attractive interaction among the π–π network among the side chains, instead of the repulsive interaction of the other amino acids in this study. An optimal balance between steric repulsion at its critical Voronoi volume and the intermolecular attractive interaction from H-bonding and van der Waals stability at the N-acyl unit presumably play a significant role in gelation self-assembly. Moreover, the present result nicely corroborates with the observation of Terech et al.,63 where it was suggested that the increase in polarophobic interaction (hydrophobic in general) from the side chains might help to increase the entanglement of the fibrous network and self-assemblies with higher conformational flexibility at the backbone architecture leading to its lower melting point as the shear elasticities, yield stress and melting behaviours correlate to the cross-sectional sizes of the fibres of the gel networks. It is also reported that β-sheets show considerably more variation in backbone dihedral angles than α-helices, leading to a variation in rotamer energy with backbone conformation,64 and the type of secondary structure of which a residue is part, in general, has a significant impact on how much side-chain entropy is lost.60 Thus, the entropic parameter on molecular packing has a significant impact over higher order molecular organization that can reflect on their three dimensional entanglement of the gelation fibres and influence the mechanical strength of the matrix. The presence and extent of elasticity in fluid gel systems were also analyzed using their tanδ values, which represent the ratio of viscous modulus (G′′) to elastic modulus (G′). The tanδ value of less than unity indicates elastic-domain (i.e. solid-like) behaviour, and values greater than unity indicate viscous-domain (i.e. liquid-like) behaviour. The tanδ values of the present set of gelators were found to be as follows 0.112 (4a), 0.118 (4b), 0.137 (4c), 0.123 (4d), 0.095 (4e) and 0.170 (4f). Based on the obtained tanδ values ranging from 0.10–0.22 in the frequency sweep measurements, the systems can be considered as strong gels, where the storage modulus G′ is on average about 6–9 times larger than the loss modulus G′′.65 The increase in the elastic modulus of an organogel, as perceived by Rogers et al., is due to an increase in fibre–fibre interactions, which lead to the formation of a greater number of transient junction zones, in turn leading to a more ordered system.66 Therefore, the elasticity (tanδ values) also corroborates with their yield stress, as well as Tgel values, in adjudicating the strength of the gels. Overall, the elasticity of the gel network decreases with the increase in the steric bulk of the amino acid side chains.
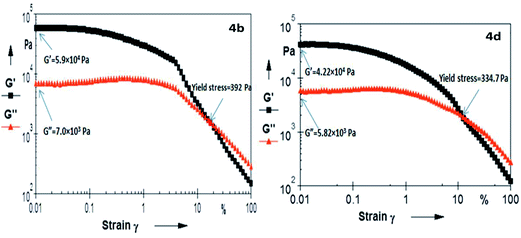 |
| | Fig. 5 Strain amplitude sweep experiments (at 10 rads per s) of gelators (4b) and (4d) in acetonitrile. | |
Wide angle X-ray powder diffraction
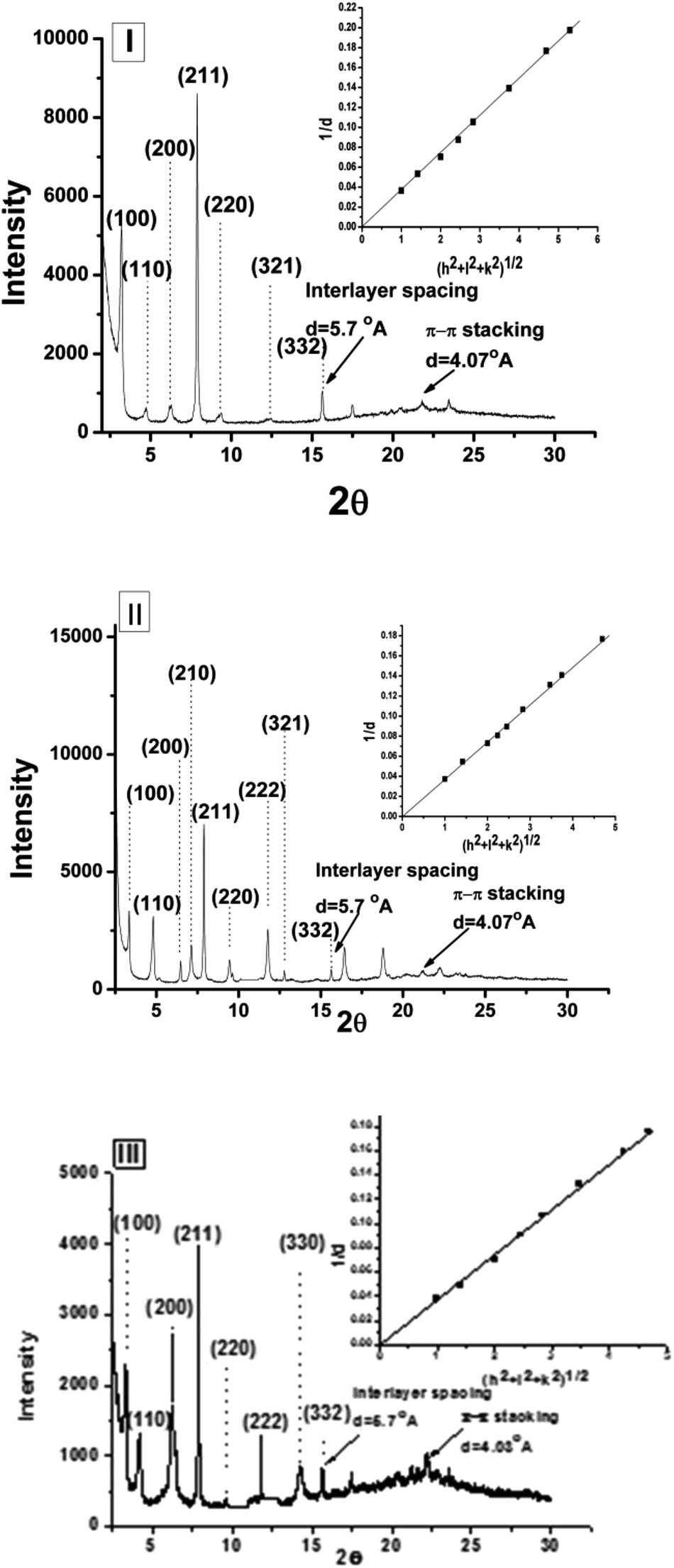
The packing pattern in the supramolecular assembly was analyzed using wide-angle X-ray diffraction (WAXD) spectra of the xerogels 4a–4f in four different solvents, of which two were aprotic (EtOAc and CH3CN) and two were protic (EtOH and 80% (v/v) EtOH–H20) (Table 3). Solvents were chosen based on (a) macroscopic non-specific solvent polarity, such as dipole moments or dielectric constant of the solvents, i.e., EtOAc (6.02), CH3CN (37.50), EtOH (24.50) and 80% (v/v) EtOH–H2O (35.72);67 (b) empirical polarity criteria, like ETN signifying the ionizing power of the solvents, EtOAc (0.228), CH3CN (0.460), EtOH (0.6540) and 80% (v/v) EtOH–H2O (0.712); between two aprotic solvents, one is comparatively apolar, i.e., EtOAc and the other one is polar, i.e., CH3CN. In general, the obtained results were found to be in good agreement with the proposed parallel β-sheet arrangement, with π–π type interactive aromatic moieties in the supramolecular network. A low-angle sharp peak with respect to the (1 0 0) plane was attributed to the depth of the self-assembly present in the molecular network and it lies between 20 to 26 Å, which is close to the extended length of a single gelator molecule. Considering the π–π type interactive aromatic units forming an extended network, keeping the molecule in one particular orientation with parallel beta sheet type H-bonded peptidic zone, the (1 0 0) reflection might be coming from the next π-electron rich aromatic layer in the opposite direction, with an interdigitated network architecture (Fig. 6), as is also evident in literature.68 Peaks assigned to a periodic distance of 5.65 ± 0.02 Å were found and were ascribed to extended two-dimensional layers of β-sheets that stack parallel on top of each other, as has been suggested earlier.29 While analyzing the diffraction pattern using the plot of 1/d vs. (h2 + k2 + l2)1/2, a straight line passing through the origin suggests that the gelation self-assembly presumably possesses a primitive cubical phase (Pm3m) with a comparable lattice parameter (a); however, intensities of some of the reflections were too low to detect. A representative plot for 4a, 4c and 4d in acetonitrile is presented in Fig. 7 (extensive plots are given in ESI, Fig. S27–S47†). The details of the major diffraction patterns (in hkl) of the gels in ethyl acetate, acetonitrile, ethanol and 80% aq. ethanol are as given in ESI.†
Table 3 The d100 spacing corresponds to first order reflection, interlayer spacing, π–π stacking and edge length of the cubic lattice (a)
| Gelators |
d100 (in Å) |
A (in Å) |
Interlayer spacing (in Å) |
π–π stacking distance (in Å) |
| 4a in EtOAc |
27.68 |
26.7 |
5.06 |
3.72 |
| 4a in CH3CN |
26.74 |
27.6 |
5.65 |
4.07 |
| 4a in EtOH |
27.75 |
26.8 |
5.67 |
3.76 |
| 4a in 80% EtOH–H2O |
27.92 |
27.7 |
5.63 |
3.80 |
|
| 4b in EtOAc |
27.40 |
27.4 |
5.06 |
3.78 |
| 4b in CH3CN |
22.57 |
22.2 |
5.68 |
3.90 |
| 4b in EtOH |
26.03 |
26.0 |
5.64 |
3.79 |
| 4b in 80% EtOH–H2O |
27.66 |
27.6 |
5.67 |
3.79 |
|
| 4c in EtOAc |
27.68 |
26.7 |
5.08 |
3.88 |
| 4c in CH3CN |
26.74 |
26.7 |
5.63 |
4.03 |
| 4c in EtOH |
26.82 |
26.8 |
5.64 |
3.89 |
| 4c in 80% EtOH–H2O |
26.67 |
26.6 |
5.64 |
3.94 |
|
| 4d in EtOAc |
26.74 |
27.6 |
5.61 |
3.89 |
| 4d in CH3CN |
26.74 |
26.7 |
5.68 |
4.17 |
| 4d in EtOH |
26.82 |
26.8 |
5.64 |
3.93 |
| 4d in 80% EtOH–H2O |
26.66 |
26.5 |
5.58 |
3.76 |
|
| 4e in EtOAc |
21.89 |
21.8 |
5.67 |
3.72 |
| 4e in CH3CN |
26.74 |
26.7 |
— |
4.01 |
| 4e in EtOH |
28.41 |
28.4 |
5.64 |
3.72 |
| 4e in 80% EtOH–H2O |
31.07 |
31.0 |
5.68 |
3.79 |
|
| 4f in EtOAc |
29.71 |
29.2 |
5.20 |
3.78 |
| 4f in CH3CN |
30.40 |
30.1 |
5.20 |
3.78 |
| 4f in EtOH |
29.71 |
29.3 |
5.21 |
3.78 |
| 4f in 80% EtOH–H2O |
30.42 |
29.9 |
5.66 |
4.03 |
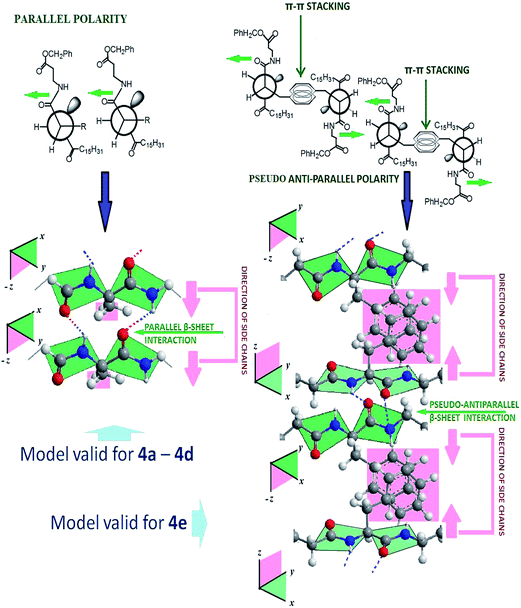 |
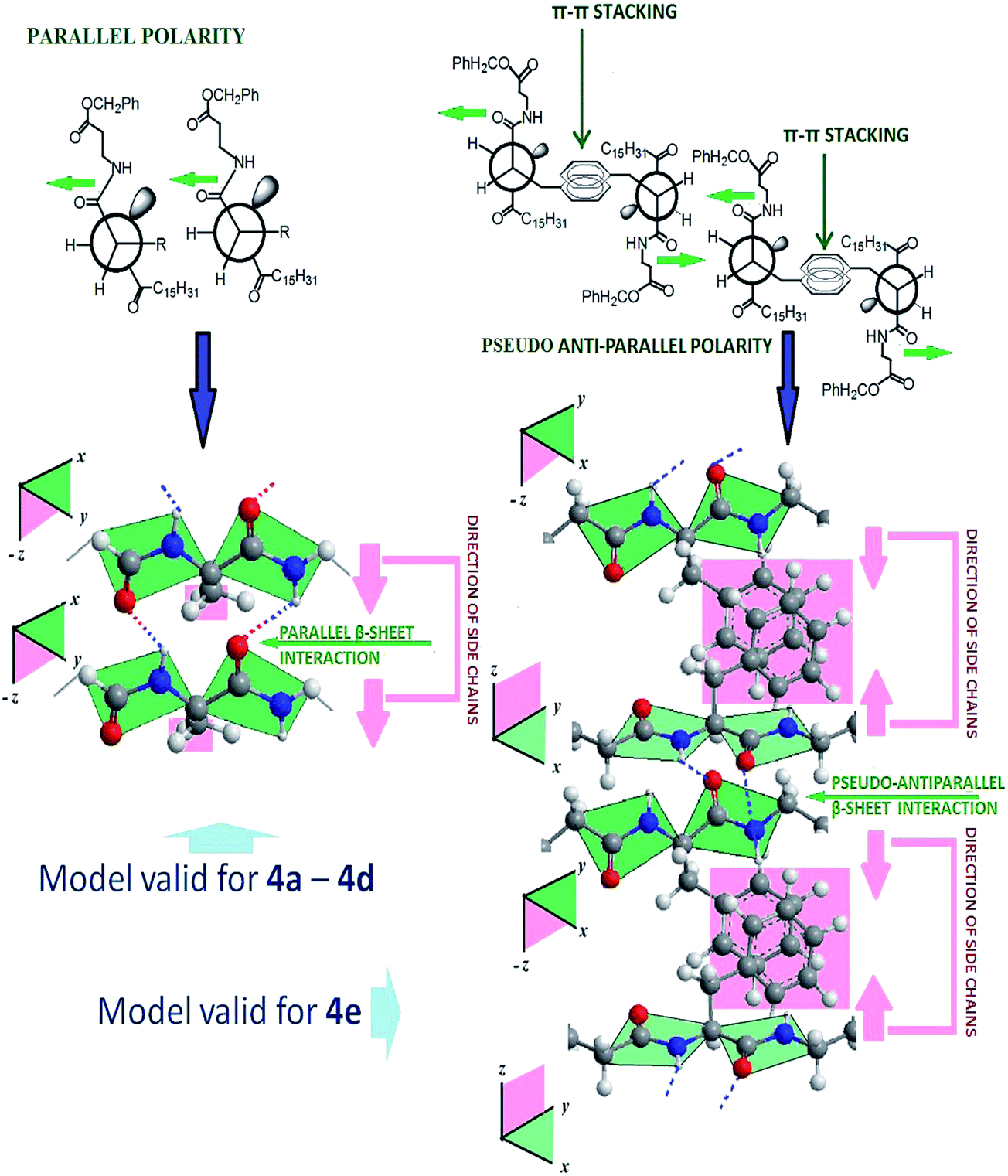
| | Fig. 6 Plausible molecular organization in the gelation self-assembly of 4a–4d and 4e. | |
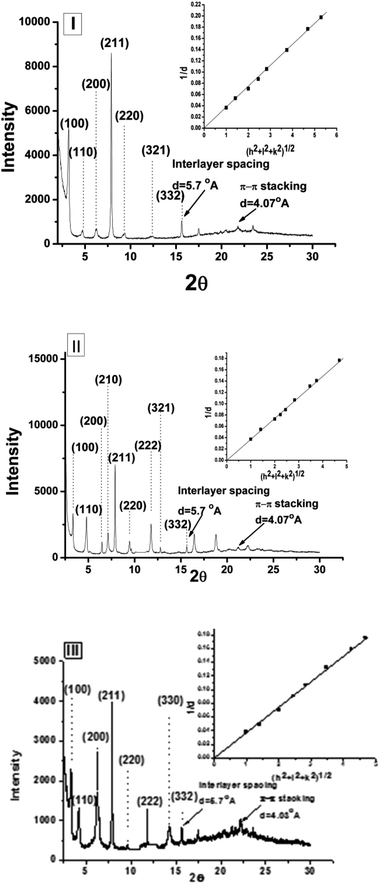 |
| | Fig. 7 XRD profile of xerogels (intensity vs. 2θ) for (I) 4a, (II) 4c and (III) 4d in acetonitrile [insets represent respective plots of d−1 vs. √(h2 + l2 + k2)]. | |
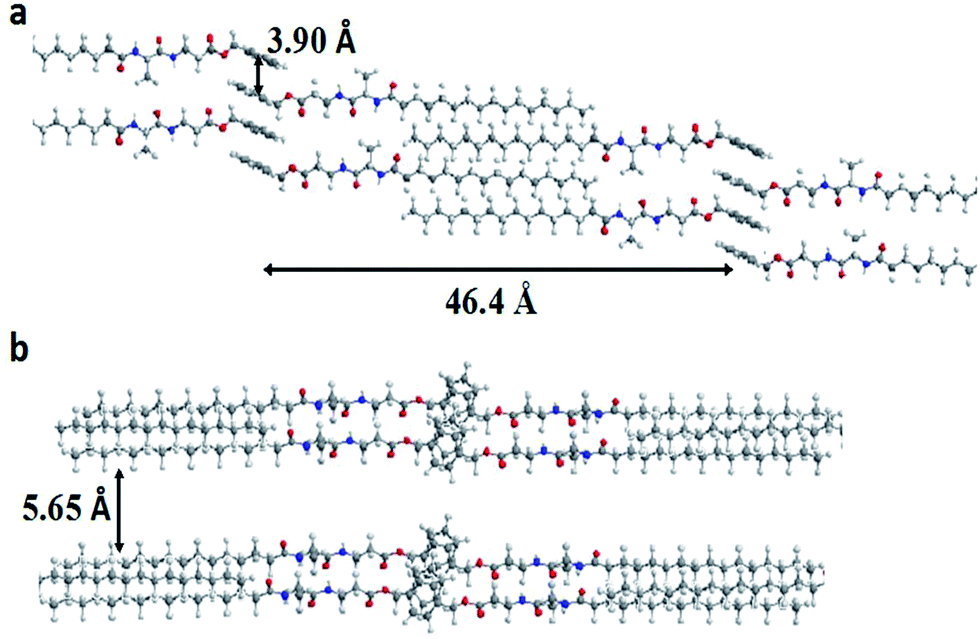
Considering that the solvent–gelator interaction is a crucial part of the gelation process, we tried to find out the effect of solvents on the packing arrangement of the gelation self-assembly, with respect to the nature of the solvents. On careful observation, we found that d100 values decrease while going from aprotic apolar to aprotic polar, and increase on going from aprotic polar to protic polar; however, it does not change much in the case of 4c. Moreover, the d100 value is higher in 80% (v/v) EtOH–H20 than in pure EtOH. Such observations clearly demonstrate that there is a substantial hydrophobic effect that helps in compact packing, while in H-bonding competing solvents (polar protic) the d100 value increases, which suggests its involvement in the amide segment that affects the molecular packing. 4c and 4d having isomeric side chains with comparable hydrophobicity, their contribution in the packing arrangement in xerogel state is found to be very similar, irrespective of the solvents. On the other hand, 4b shows the lowest d100 value in MeCN, signifying its better packing arrangement and a higher d100 value for 4f with weaker packing; however, this value remains remarkably identical for all other gelators under investigation. Surprisingly, 4f shows comparatively a very high lattice parameter (a) and this can be accounted for by the extra void space as observed with the Voronoi volume.42 Likewise, a similar lattice parameter for 4c and 4d can also be attributed to their comparable Voronoi volume and their hydrophobicity. Based on the same parameter, a lower Voronoi volume for 4a must give a lower lattice parameter than 4b, which is applicable in apolar solvents like EtOAc, but in polar media (in CH3CN or in EtOH), the lattice parameter of 4b is comparatively lower than 4a. This could be due to the inherent conformational flexibility as the isentropic pressure coefficient of Val (1.8) < Ala (3.7). Moreover, the hydrophobicity of Val is higher than Ala and that could play a role in polar media. Therefore, the fine balance between Voronoi volume, substituents conformational flexibility (equated to its isentropic pressure coefficient) and the hydrophobic factor leads to the optimal packing arrangement of the self-assembly. The obtained results also suggest that an interdigitated self-assembly might be prevalent in the molecular arrangements as the d-spacing is shortest in the case of 4b (22.57 Å) and longest in 4f (30.4 Å); however, the d100 and interlayer-spacing values are surprisingly identical for all other cases (26.74 Å) in CH3CN. Using molecular mechanics (CS Chem3D ultra version 7.0), the molecular length of 4b was calculated and a gelator self-assembly was made for molecular mechanics energy minimization, which suggests considerable interdigitation among the acyl chains with d100 value calculated to be ∼46.4 Å, however d100 obtained by XRD measurements is 22.57 Å, suggesting that the interdigitated self-assembly might have a tilt angle of ∼60° (Fig. 8). The π–π stacking distance was also found to lie within the range 3.9–4.1 Å, which is consistent with our earlier results.37
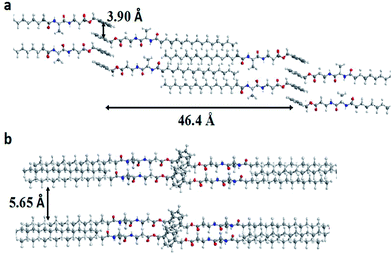 |
| | Fig. 8 A representative self-assembly of the gelator 4a: (a) interdigitated acyl chains with a parallel β-sheet type arrangement with a calculated d100 value and π–π stacking distance among aromatic groups and (b) showing the inter layer spacing. | |
UV-Vis and fluorescence spectroscopic studies
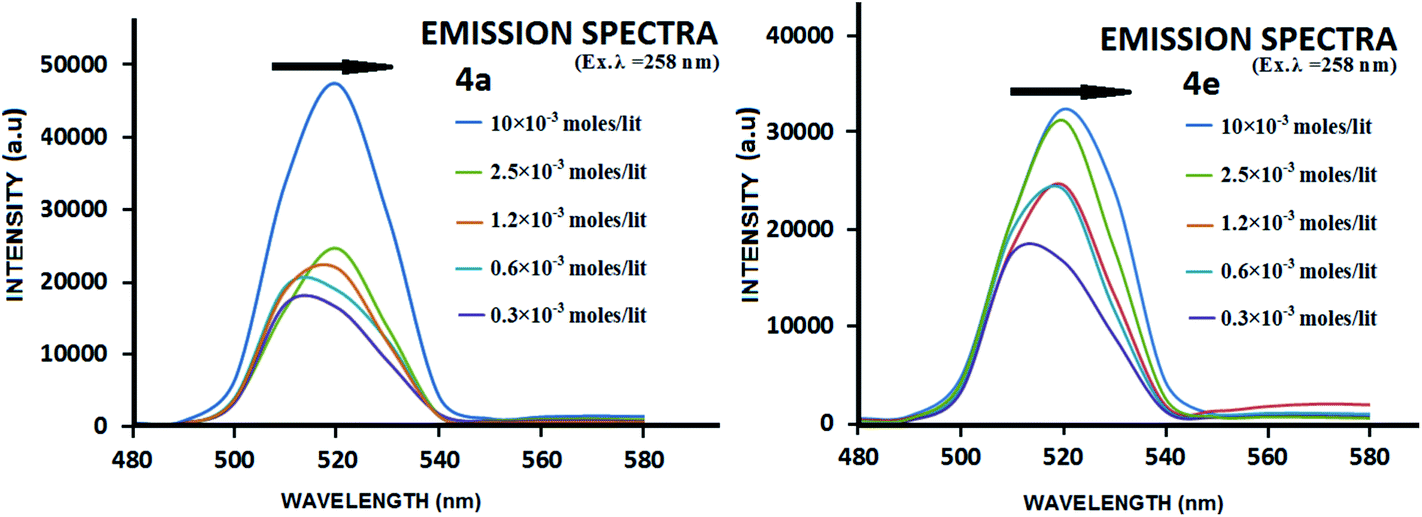
The presence of π-stacking interactions in gelation self-assembly has been witnessed in WAXRD; it was also checked by UV-Vis and fluorescence spectroscopic studies. In general, the preferred orientation of π-stacked aromatic units occurs in two modes: J- and H-type aggregation,69 which depend on the tilt angle (θ) between the molecular axis and the transition dipole moment of the chromophore. J- and H-type aggregation presumably form when θ < 54.7° and θ > 54.7°, respectively. A bathochromic shift of the monomeric band of the chromophore in the UV-Vis spectra is attributed to the J-type aggregate, whereas a hypsochromic shift is assigned for the H-type aggregates. In the present study, the UV-Vis spectra did not show a significant shift (shown in ESI – Fig. S48–S50†) with the increase in gelator concentration, which led us to record the excitation spectra of the same. While doing so, we found that the usual emission wavelength (∼285 nm) of phenylalanine (at λex = 260 nm) was not significant, while a strong band appeared at 520 nm, which was red shifted with the increase in concentration of the gelator (Fig. 9). Such red shifted emission (509–520 nm with the increase in concentration from 0.3 mM to 10 mM) for a fixed excitation wavelength also indicates clearly that this emission is not due to the overtone of the excitation, rather such concentration dependent red shifted emission at ∼520 nm of phenyl units is corroborated to the earlier results by various groups,70 which is attributed to the excimer emission of the π-stacked phenyl rings in the gelation self-assembly. The red shift in such emission has been ascribed to face-to-face organization of the π-stacked aromatic units.71 To check the appearance of the band at 520 nm as an eximer band of 260 nm, we have recorded the excitation spectra with λ = 520 nm which gives the absorption band at 260 nm (given in ESI Fig. S50†).
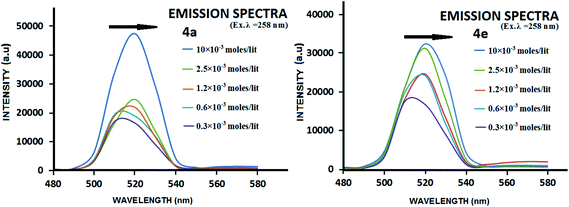 |
| | Fig. 9 Fluorescence emission spectra of the gelators 4a and 4e at different concentrations in acetonitrile. | |
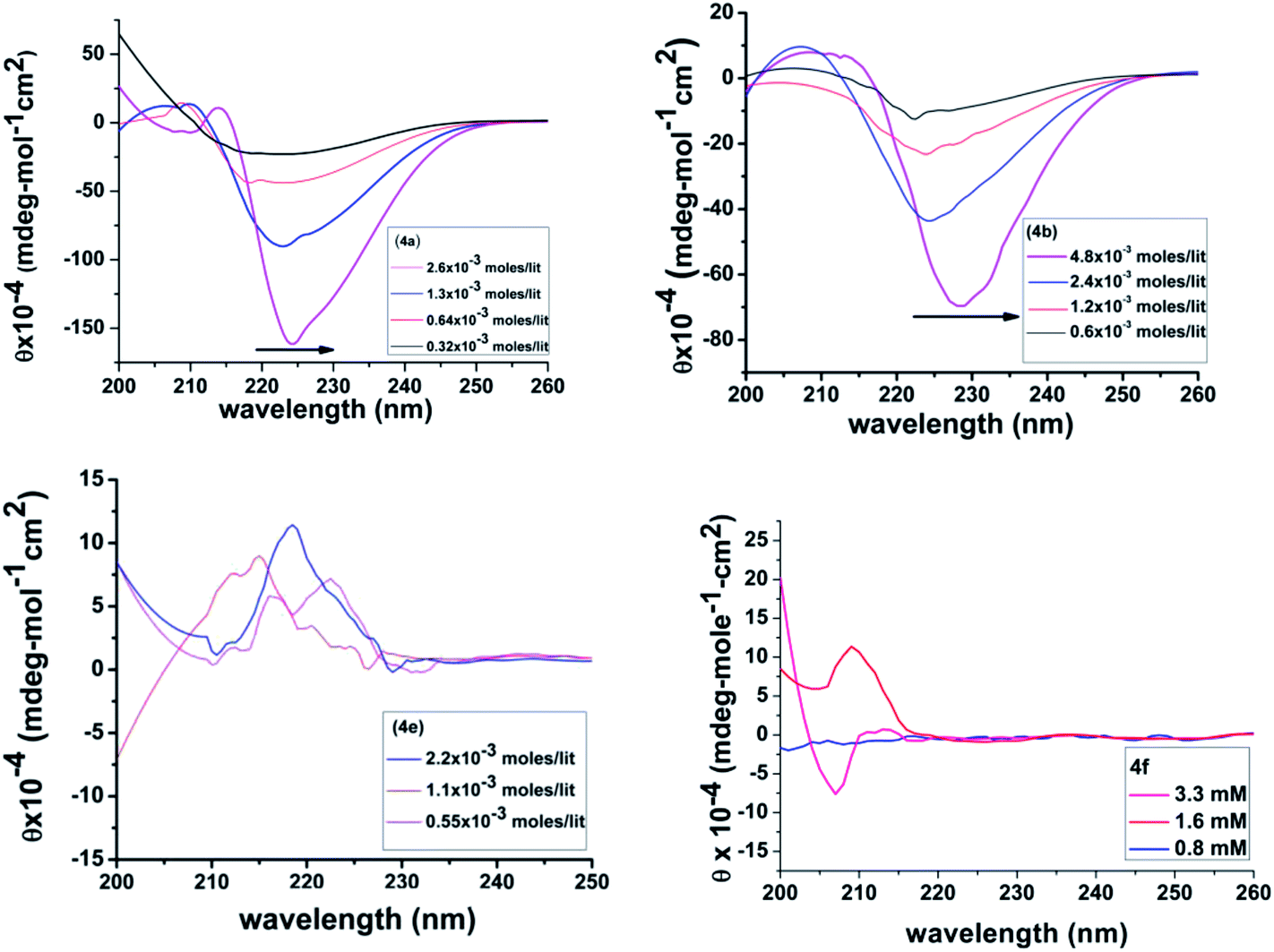
Circular dichroism
Nucleation → growth → fibre formation has been found to be a part of the gelation self-assembly. The initial formation of a one dimensional supramolecular network via self-assembly of gelator molecules was found to be an important event for the generation of 3D entangled fibrils in the gelation process. To understand such an event, a concentration dependent circular dichroism study was performed for the present set of gelators. To avoid scattering, experiments were carried out at a concentration much lower than their corresponding CGC values. In the present study, CD signatures were obtained at three different concentrations, 0.55 mM, 1.1 mM and at 2.2 mM (Fig. 10). In general, all the gelators showed very similar, strong, positive couplets in the CD spectra, except 4e with a higher wavelength (n → π*) negative Cotton band at ∼238 nm and a positive Cotton band in the lower wavelength region (π → π*). In the case of 4e, a distinctly different positive Cotton band was observed, which also had different signatures with the increase in concentration. For 4a–4c, the red shift was clearly identified in the CD spectra for the higher wavelength n → π* band with the increase in concentration of the gelator. Such a shift can be attributed to the fact that a considerable deviation of the planarity of the amide functionality in the self-assembly occurs at higher concentration with the advent of nucleation for the gelation. Such an observation was nicely described by Bednárová et al. in their computational studies on the spectroscopic properties of nonplanar amide groups.72 Therefore, such phenomena suggest that a significant conformational change on the gelator backbone is associated with the formation of a gel network at higher concentration. Unequal amounts of red-shifts of the CD bands at higher wavelengths for different gelators also suggest that the gelator backbone is identical in all cases; the conformational flexibility of the side chain segments differently influence the gelator self-assembly.73 The distinct behaviour of phenylalanine having a positive Cotton band is quite common in the literature.74 Herein, in 4e, a small positive Cotton band at 212 nm and a probable negative band below 200 nm might signify the self-assembly in a random coil architecture at a concentration of 0.55 mM. However, at a concentration of 1.1 mM, the previous negative band disappears and instead, positive Cotton bands at 212 nm (lower intensity), 216 nm and 222 nm are observed, which indicate a more ordered structure of the self-assembly. These bands are attributed to the La band of phenylalanine that originates from the rotational strength through coupling with nearest neighbour peptide groups,75 where both the 1A1g → 1B1u transition of the phenylalanine ring and the π–π* transition of the peptide chromophore contribute to the optical activity.76 The advent of a strong positive Cotton band near 220 nm, as observed for the 2.2 mM concentration is attributed to the π–π stacking interaction of the aromatic side chains of phenylalanine, as corroborated by the results obtained by Hamley and co-workers in the self-assembly of model amphiphilic phenylalanine peptides.77 The molecular organization with π–π stacking of the 4e gelator (Fig. 6) results in a centrosymmetrically placed, specific molecular orientation having a better π–π interaction at the aromatic sides, orthogonally placed with respect to the amide plane of the parallel β-sheet organization. This leads to a pseudo-antiparallel H-bonding interaction between amide groups facing opposite to the side chains becoming plausible, which is supported by our previous observation of phenylalanine based surfactant self-assembly32 and was later substantiated by Francesc Sagués et al., confirming that possible π–π interactions between the aromatic groups of 8-Az3COOH and L-Phe may occur, giving rise to non-birefringent aggregates.78 The existence of such an antiparallel effect was also evident in IR studies [see IR studies].
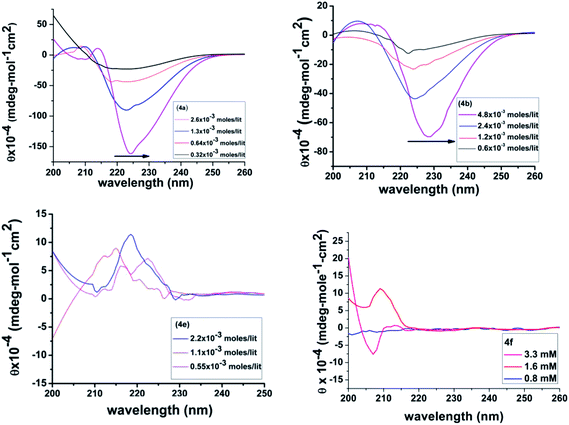 |
| | Fig. 10 Concentration dependent CD spectra of 4a, 4b, 4e and 4f in CH3CN. | |
To understand the role of chirality in gelation self-assembly, concentration dependent CD studies were also performed, with 4f having an achiral glycine residue. A very interesting result was observed while recording the CD spectra from a low to high concentration. At a very low concentration (∼0.8 mM) there was no Cotton band but at a concentration of 1.6 mM, a positive Cotton band at wavelength of 209 nm appeared and at a moderately high concentration at 3.2 mM, a negative Cotton band at 207 nm started to appear. Such a CD signature from an achiral molecule does implicate the advent of an extended H-bonding network with a β-sheet type arrangement, with the growth of the self-assembly upon increase in concentration. Thus, the results strongly suggest that chirality might not be the sole prerequisite for gelation self-assembly; it is the proper alignment of supramolecularly interactive moieties of the molecule and solvophobic association that play a dominant role.
FT-IR studies
The inter-gelator interaction at the peptidic region and the ester functionality near the aromatic substituent have been investigated by FT-IR studies, by identifying the changes in the vibrational spectra of the non-covalently interactive functional units in the gel and sol states of the gelators. Since amide-I and amide-II bands are the two most prominent vibrational bands to identify protein secondary structures, we tried to analyse the accompanied band shift of non H-bonded amide in the soluble state to the H-bonded amide in the gel state as identified by FT-IR. The analysis of the FT-IR spectra of gelators (4a–4f) was performed in solution (CHCl3), as well as in the gel state in acetonitrile (shown in Table 4) (ESI, Fig. S9–S14†). The amide I vibration, (absorbing near 1635 cm−1) arises primarily from the C–O stretching vibration, along with minor contributions from the out-of-phase C–N stretching vibration, the C–C–N deformation and the in plane bending of NH. In the present case, all the systems show absorptions within the range of 1630–1643 cm−1, which lies within the characteristic amide-I band for parallel β-sheet type amide networks. Moreover, the amide-II band within the range of 1540–1547 cm−1 also supports the parallel β-sheet type arrangement. Usually, the amide II band arises from the out-of-phase combination of the NH in plane bend and the CN stretching vibration with smaller contributions from the CO in plane bend and the C–C and N–C stretching vibrations. While comparing the data for 4e to others, a striking difference was observed. Additional absorption bands at 1698 cm−1 and 1510 cm−1 (along with 1637 and 1538 cm−1) were observed, suggesting that a mixed β-sheet type peptidic network (anti-parallel and parallel) exists in the gel network. Primarily, an invariant band at 1738 cm−1, assigned to ester C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, across all the gelators at their gel or sol state suggests its non-participation in H-bonding.79 To put it in a simpler perspective, two types of molecular organizations have been postulated, which are depicted in Fig. 6. Since the side chain residues remain orthogonal to the H-bonded plane of the parallel β-sheet organization, for minimizing the inter side chain repulsive interaction for 4a–4d, the side chains place themselves as far apart as possible, which likely leads to a parallel β-sheet type molecular organization on both longitudinal and transverse sides of the amide planes. The same is apparently true for the achiral 4f. However, in the case of 4e, to optimize the π–π type attractive interaction from the aromatic side chains, two parallel β-sheet planes in the longitudinal direction might stack in a face to face centrosymmetric fashion, leading to one of the β-sheet faces being organized into a pseudo-antiparallel fashion from the neighbouring plane. Such organization is presumed to be unique for the 4e system, having such π–π interactions of the side chains. Such organization was further demonstrated in CD studies as well (see in CD study). Similar d100 values in XRD data further suggest that such mixed orientation does not affect the interdigitation picture of the gelation self-assembly much.
O, across all the gelators at their gel or sol state suggests its non-participation in H-bonding.79 To put it in a simpler perspective, two types of molecular organizations have been postulated, which are depicted in Fig. 6. Since the side chain residues remain orthogonal to the H-bonded plane of the parallel β-sheet organization, for minimizing the inter side chain repulsive interaction for 4a–4d, the side chains place themselves as far apart as possible, which likely leads to a parallel β-sheet type molecular organization on both longitudinal and transverse sides of the amide planes. The same is apparently true for the achiral 4f. However, in the case of 4e, to optimize the π–π type attractive interaction from the aromatic side chains, two parallel β-sheet planes in the longitudinal direction might stack in a face to face centrosymmetric fashion, leading to one of the β-sheet faces being organized into a pseudo-antiparallel fashion from the neighbouring plane. Such organization is presumed to be unique for the 4e system, having such π–π interactions of the side chains. Such organization was further demonstrated in CD studies as well (see in CD study). Similar d100 values in XRD data further suggest that such mixed orientation does not affect the interdigitation picture of the gelation self-assembly much.
Table 4 Selected FTIR bands in wave numbers [cm−1] for the gelators in acetonitrile (in the gel state) and chloroform (in solution state)
| |
N–H stretching (cm−1) |
Amide II (cm−1) |
Amide I (cm−1) |
Ester (cm−1) |
| Sol |
Gel |
Sol |
Gel |
Sol |
Gel |
Sol |
Gel |
| 4a |
3430 |
3292 |
1522 |
1541 |
1664 |
1636 |
1737 |
1733 |
| 4b |
3433 |
3286 |
1527 |
1534 |
1664 |
1632 |
1730 |
1730 |
| 4c |
3433 |
3297 |
1530 |
1541 |
1667 |
1634 |
1735 |
1734 |
| 4d |
3444 |
3240 |
1531 |
1541 |
1664 |
1636 |
1737 |
1733 |
| 4e |
3436 |
3302 |
1521 |
1538 |
1665 |
1637 |
1740 |
1738 |
| |
|
|
|
1510 |
|
1698 |
|
|
| 4f |
3321 |
3219 |
1520 |
1548 |
1667 |
1636 |
1725 |
1725 |
Solvent effect
Residing at the critical domain between two extremes of solvent–solute interactions leading to solubility or insolubility, the solvent plays a vital role in the advent of gelation self-assembly, its performance and properties.53 Solvent effect on gelation has majorly been highlighted based on solvent–gelator interaction at its macroscopic level using different solubility parameters of the solvents related to its nonspecific and specific intermolecular forces.80 Although the gelator–gelator interactions have been studied by various research groups, the additional contribution from solvent–solvent interaction has not been addressed clearly towards tuning the gelation behaviour along with the gelator architecture. Between the two distinct classes of intermolecular forces, the first category is comprised of the so-called non-directional, induction and dispersion forces, which are non-specific and cannot be completely saturated, whereas the other category consists of specific and directional forces like hydrogen-bonding forces, charge-transfer or electron-pair donor–acceptor forces or π–π stacking interactions, which can be saturated and lead to stoichiometric molecular compounds. Dordick and his co-workers reported that gelation number decreases with the increase in the Hildebrand solubility parameter for the same class of solvents for trehalose-6,6′-diacetate and 6,6′-dibutyrate.81 The minimum gelation concentration of cyclo-(dipeptides) in various organic solvents was correlated with a polar solubility parameter (δa) of the solvents by Hanabusa and co-workers.82 The general relationship between macroscopic gelation and solvatochromic parameters, based on a multi-parameter approach to solvent polarity using Kamlet–Taft parameters were reported by Smith and co-workers.83,84 A linear correlation between Tgel values and the dielectric constants of alcoholic solvents were reported by Makarević and co-workers for the gelation of different bis(amino acid)oxalyl amides.85 H-Bonding interactions were found to be weaker in polar solvents, and gelation occurred only where sufficient compensation was provided by intergelator van der Waals interactions in cyclohexane based bisamide and bisurea organogelators, as highlighted by Zweep et al.86 Solvophobic interactions towards peptide based organogelation was also addressed by Loo et al.; describing this interaction involves the solvent molecules, as well as solvophobic groups in the interior of the aggregate.87 Herein, we have tried to discuss the gelation ability of the gelators 4a–4f in different solvents based on different solubility parameters:88 (a) Dimroth–Reichardt ETN (normalized ET(30) parameter), (b) Hansen–Beerbower solubility parameters (HSP), (c) Kamlet–Taft parameters. It is well understood in the literature, with lots of reports that solvent–gelator and gelator–gelator interactions are important, and we do believe in that as well. In 1979, Professor J. H. Hildebrand of University of California at Berkeley did a critical comment on hydrophobicity and he said: “In conclusion, there is no hydrophobia between water and alkanes; there only is not enough hydrophilia to pry apart the hydrogen bonds of water so that the alkanes can go into solution without assistance from attached polar groups.”89 Despite extensive research devoted to the hydrophobic effect, its molecular mechanisms remain controversial.90 As the gelation phenomena is majorly influenced by solvophobic interaction, it is envisaged that the solvent–solvent cohesive interactions can also be considered to look at its effect towards the gelation process. Gordon parameters, which deal with the cohesiveness of solvents, correlate with the solvent–gelator interaction and the obtained results are presented to check whether it has any role in the gelation process. The mechanism for the self-assembly process of an amphiphile in organic solvents has accounted for the solvophobic effect, which is linked to the solvent cohesiveness as quantified by the Gordon parameters by Greaves et al.91 Similarly, herein we have tried to correlate the gelation behaviour with the cohesiveness of the gelating solvents where the cohesiveness is represented by their Gordon parameter (G = γ/Vm1/3); where γ is the surface tension and Vm is the molar volume.92 The trend shows some dependence and we feel that such cohesive interactions among solvent molecules might not be the sole reason; however, they cannot be overlooked. The ETN parameter majorly signifies the ionizing power (polarity in a simpler sense) of the solvents. On the contrary, HSP comprises of three components (i) energy from dispersion forces involved between the molecules (δd), (ii) energy from dipolar intermolecular forces (δp) and (iii) energy from hydrogen bonds between molecules (δh). The δo and δa are the total solubility parameter and the polar solubility parameter, respectively. The total solubility parameter δo has been described as δo2 = δd2 + δa2, where δa2 = δp2 + δh2. For small molecules, (δo) is equated to the cohesive energy that is required to overcome the intermolecular forces. Moreover, parameters of the Kamlet–Taft solvatochromic relationship, which measures separately the hydrogen-bond donor (α), hydrogen bond acceptor (β), and dipolarity/polarizability (π*) properties of solvents, contribute to overall solvent polarity. These usually relate to the overall solvation ability for the solutes at the ground state/and their excited states. The role of solvents on any particular gelator has been analyzed with the data over Hansen space, Teas plot and separately, with the GN versus different solvent parameters. The Hansen space and Teas plot have been scrutinized (shown in ESI, Fig. S15–S21†) to identify the prevalence of the solvents that support the gelation process. The analysis describes how solvents with higher δd and lower δh of HSP and lower α of KT parameters help the gelation process. This suggests that solvents with H-bonding donating ability hinder the gelation, whereas solvents with higher dispersive forces augment the gelation phenomena. Such analysis was also corroborated by the findings that the solvents with higher δp (HSP)/ETN/Gordon parameters show better gelation ability as found by their GNs in Fig. 11 (ESI, Fig. S15–S26†). This suggests that the polarophobic effect, as well as the cohesive nature of solvents (higher Gordon parameter) play a significant role in the gelation process. This has also been substantiated by the similar trend of gelation behaviour in protic polar solvents like alcohols. Moreover, the solvent molecule architecture plays a significant role, as the weakest gelation was observed in isopropanol, compared to n-butanol. We also observed that the chemical nature of the solvents play a major role, even with comparable solvent parameters. EtOH and nitrobenzene have comparable δp (HSP) of 8.8 and 8.6, respectively, but 4a shows gelation in ethanol but not in nitrobenzene. Similarly, 1,2-dichlorobenzene (DCB) and ethylacetate (EtOAc) have comparable ETN values of 0.225 and 0.228, respectively; however, 4b shows much better gelation in EtOAc, compared to DCB. In the case of Gordon parameters as well, toluene and ethanol have quite comparable values of 0.588 and 0.566, respectively; the gelation ability of 4a/4b/4c is less in toluene than in EtOH. Based on the trend of the obtained results, the solvents under investigation were segregated and on a closer look it was seen that they can be clustered based on their significantly different polarizability (π*) values of Kamlet–Taft solubility parameters, such as the aromatic solvents, into two categories: (i) with aliphatic substituents (benzene, toluene and o-xylene) with 0.51 < π* < 0.59 and (ii) with electronegative substituents (anisole, 1,2-dichlorobenzene and nitrobenzene) with 0.73 < π* < 1.01. Similarly, we divided the polar aprotic ones into two groups: (i) EtOAc, acetone and acetonitrile with 0.55 < π*< 0.75 and (ii) DMF, DMSO and nitromethane with 0.85 < π* < 1.0. The alcoholic solvents were put as a single class of polar
protic solvents, where 0.47 < π* < 0.6. When we compare their δp, ETN and the Gordon parameters with gelation numbers, it shows an increasing trend of gelation number with the increase of the values of the respective parameters. The gelation in protic polar solvents, i.e., in alcohols, meaningfully indicates that H-bonding interactions among solvents and gelators were unable to destroy the amide network of gelator–gelator interactions. These results suggest that while comparing the gelation behaviour of the solvents, not just the numerical values of the corresponding solubility parameters, the chemical nature of the solvents, as distinguished through their polarizability values (π* of KT solubility parameter) should be considered as well. However, a question still remains: what are the forces helping in the solubilisation of the gelators in the media? A plausible answer to that is the architectural functionalities that weaken the intergelator packing by increasing the intergelator distance, mainly by repulsive interaction (from the side chains of amino acids for the present systems), the increase in solvent–solute interaction via various non-covalent forces, and last but not least, there might be a weaker cohesive force working in solvent–solvent interactions with a reduced unfavorable entropy factor. Therefore, a cooperative optimization is indispensable for gelation self-assembly.
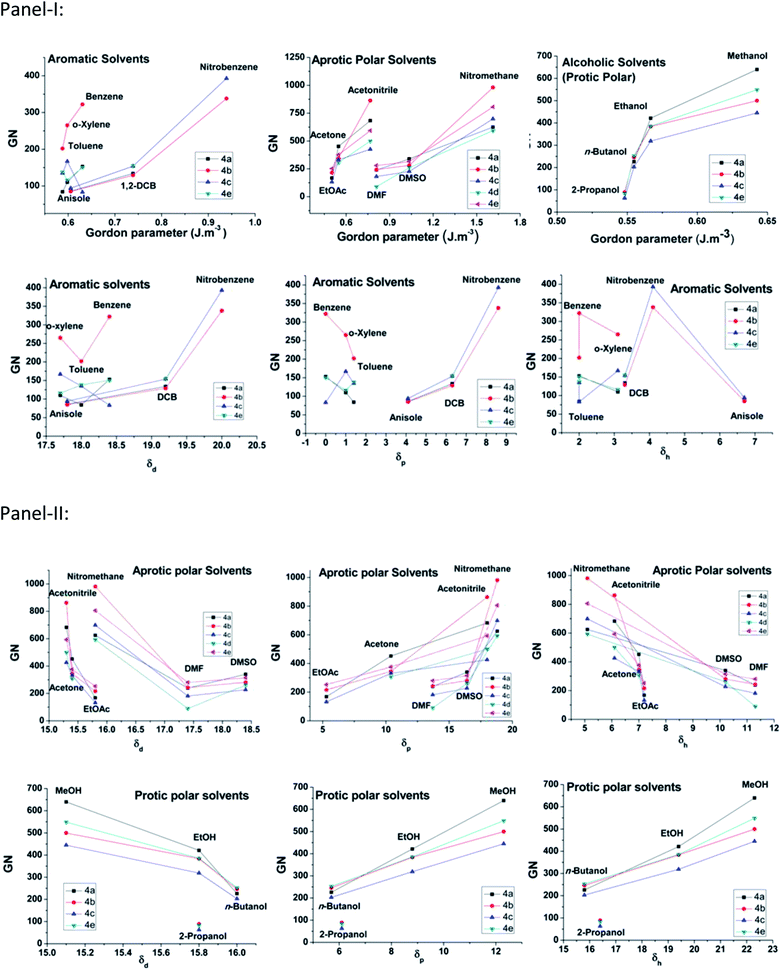 |
| | Fig. 11 Panel-I represents the plot of GN versus Gordon parameter and GN versus HSP; panel-II represents the plot of GN versus HSP in segregated solvent systems as indicated. | |
Conclusion
To understand the role of amino acids in a self-assembly, a rational approach is always required to have a proper structure–function relationship. Herein, one such rational approach has been taken, with a defined complexity that has not been addressed earlier. A fine tuning of supramolecular ambidextrous gelation properties in apolar and polar organic media (including aqueous alcohol mixtures) has been accomplished by altering an encrypted α-amino acid with various hydrophobic side chains on a designed gelator backbone architecture with a common formula: N-n-hexadecanoyl-αAA-(β)Ala-OBn (α-AA = L-isomers of Ala (4a), Val (4b), Leu (4c), Ile (4d), Phe (4e) and Gly (4f)). In addition to the supramolecular interactions, including H-bonding through amide segments, three important parameters related to side chains have been found to be decisive in rationalizing the obtained results: (i) hydrophobic effect of the amino acids (measured by hydrophobicity scale), (ii) van der Waals type steric repulsions (analyzed based on the Voronoi volume of the residues) and (iii) the flexibility of the side chains (conformational flexibility scale). A cooperative interaction involving (a) hydrophobic effect, (b) steric repulsion among the side chains, (c) its volume at its compact organization, (d) the conformational flexibility arising from optimal reorientation at the side chains with a reorganization of gelator backbone via non-planarity at the amide segments, seems to play a fundamental role in dictating the gelation properties of the gelators. In general, an optimal balance among all the above parameters becomes apparently ideal for 4b ≥ 4d > 4a > 4c. Between 4c and 4d with isomeric side chains, 4c shows much weaker gelation, whereas, GN is found to be much lower for 4d, compared to 4b, even with more hydrophobicity in 4d compared to 4b with an extra Cγ-methyl substitution. Thus, hydrophobicity cannot be the sole reason for the difference in gelation ability among the gelators. This could be due to the difference in molecular packing arising from optimal side chain volume and its inherent conformational flexibility. 4a with the least substituted chiral amino acid with methyl group also shows good gelation behaviour, but not with the entire range of solvents, compared to 4b or 4d. Between 4c and 4d, with isomeric side chains, 4c shows weaker gelation, presumably due to its higher side chain conformational flexibility; nevertheless, their contribution in the packing arrangement in the xerogel state is found to be very similar, irrespective of the solvents. Interestingly, 4e gets the extra stability due to its side chain π–π type attractive interaction with a pseudocentro-symmetric H-bonded amide network, compared to the repulsive type interaction that exists for others. The WAXRD data also reveal that d100 values decrease while going from aprotic apolar to aprotic polar, while it increases on going from aprotic polar to protic polar media, demonstrating that a substantial hydrophobic effect helps in compact packing, whereas in H-bonding competing solvents (polar protic) the d100 values increase, attributed to the involvement of solvents with the amide segment, which affects the molecular packing. The role of solvents in such amino acid modulated self-assembly was also addressed using Hansen solubility parameters (HSP), Demorth–Reichardt ETN parameter, Kamlet–Taft solubility parameter (KT) and Gordon parameter of the solvents. In addition to solvent–gelator and gelator–gelator interactions, solvent–solvent cohesive interaction (presented by Gordon parameters) was also taken into account. The polarizability parameter of KT was found to have a distinct effect for solvents having very similar ETN or HSP values, signifying the important polarophobic effect for gelation self-assembly. The analysis of obtained data also accounts for the unique molecular arrangement with a peptidic motif having a single amino acid placed in a parallel β-sheet type orientation. Comparing the GN values in nitromethane (aprotic polar) and 80% EtOH–H2O, it can be visualized that the hydrophobic complementarity of side chains follows the trend 4b > 4e > 4d > 4c > 4a > 4f in the former, whereas, in protic polar media a suspected interaction of solvents on the H-bonding amide segment of the overall hydrophobic collapse would lead to better packing, even with lower bulk substituents and the trend becomes 4a > 4b > 4d > 4e ≈ 4c > 4f in 80% EtOH–H2O. An apparently better gelation of 4b and 4d can also be attributed to their higher propensity for β-sheet stabilization in protein chemistry. Therefore, as a whole, a polarophobic association with a synergic support of optimum supramolecular interaction involving a cooperative effect arising from a critical balance of side chain hydrophobicity, volume of steric bulk at optimal packing and its inherent conformational flexibility of the inherent spatial geometry with the required reorientation at the flexible and/or apparently rigid segments, play a crucial role in dispersive and directional forces operating behind such tuning of the physicochemical behaviour of the self-assembly system. We do believe that such studies on the structure–activity relationship of amino acid derived supramolecular self-assembly systems can immensely help towards the de novo design of amino acid based, next generation nanostructured soft-materials with controllable properties.
Acknowledgements
Financial support from Department of Science and Technology (DST) (Grant No. SR/S1/OC-67/2008), Govt. of India and UPE-II (under UGC), Jadavpur University. Authors also thank Mr Suman Halder and Mr Sudipta Mukherjee of UPE-II for their help in performing rheological experiments. UPE-II is also acknowledged by KK for her research fellowship.
References
- A. Hirst, S. Roy, M. Arora, A. K. Das, N. Hodson, P. Murray, S. Marshall, N. Javid, J. Sefcik, J. Boekhoven, J. van Esch, S. Santabarbara, N. Hunt and R. Ulijn, Nat. Chem., 2010, 2, 1089–1094 CrossRef CAS PubMed.
- P. Beseniusa, G. Portaleb, P. H. H. Bomansc, H. M. Janssend, A. R. A. Palmansa and E. W. Meijer, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 17888–17893 CrossRef PubMed.
- J. N. Israelachvili, Intermolecular and Surface forces, Elsevier, 3rd edn, 2011, ch. 20, p. 535 Search PubMed.
- R. Weiss, J. Am. Chem. Soc., 2014, 136, 7519–7530 CrossRef CAS PubMed.
- P. Koehl and M. Levitt, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 12524–12529 CrossRef CAS.
- Y. Hong, R. Legge, S. Zhang and P. Chen, Biomacromolecules, 2003, 4, 1433–1442 CrossRef CAS PubMed.
- A. Hirst, I. Coates, T. Boucheteau, J. Miravet, B. Escuder, V. Castelletto, I. Hamley and D. Smith, J. Am. Chem. Soc., 2008, 130, 9113–9121 CrossRef CAS PubMed.
- R. Fairman and K. Åkerfelgt, Curr. Opin. Struct. Biol., 2005, 15, 453–463 CrossRef CAS PubMed.
- A. Vintiloiu and J. Leroux, J. Controlled Release, 2008, 125, 179–192 CrossRef CAS PubMed.
- S. Zhang, X. Zhao and L. Spirio, Scaffolding in Tissue Engineering, ed. Peter X. Ma and Jennifer Elisseeff, CRC Press, Taylor and Francis group, 2006, p. 217 Search PubMed.
- G. John, S. Jadhav, V. Menon and V. John, Angew. Chem., Int. Ed., 2012, 51, 1760–1762 CrossRef CAS PubMed.
- M. Ni and C. A. E. Hauser, Micro and Nanofabrication Using Self-Assembled Biological Nanostructures, ed. J. Castillo-León and W. E. Svendsen, Elsevier, 2015, ch. 4, p. 64 Search PubMed.
- J. Zhang and C. Su, Coord. Chem. Rev., 2013, 257, 1373–1408 CrossRef CAS.
- I. Chernynad and E. Gazit, Angew. Chem., Int. Ed., 2008, 47, 4062–4069 CrossRef PubMed.
- F. Fages, Angew. Chem., Int. Ed., 2006, 45, 1680–1682 CrossRef CAS PubMed.
- J. Lee, J. Park, J. Park, H. Ahn, J. Jaworski and J. Jung, Nat. Commun., 2015, 1–9 Search PubMed.
- A. Bui and N. Virgilio, Ind. Eng. Chem. Res., 2013, 52, 14185–14191 CrossRef CAS.
- J. Lu, J. Wua and Y. Ju, New J. Chem., 2014, 38, 6050–6056 RSC.
- S. Abraham, R. Vijayaraghavan and S. Das, Langmuir, 2009, 25, 8507 CrossRef CAS PubMed.
- J. Bachl, J. Mayr, F. Sayago, C. Cativielab and D. Díaz Díaz, Chem. Commun., 2015, 51, 5294–5297 RSC.
- C. Tang, R. V. Ulijn and A. Saiani, Langmuir, 2011, 27, 14438–14449 CrossRef CAS PubMed.
- S. Han, Chem.–Eur. J., 2011, 11, 13095–13102 CrossRef PubMed.
- A. Marchut and C. Hall, Biophys. J., 2006, 90, 4574–4584 CrossRef CAS PubMed.
- C. Dobson, Methods, 2004, 34, 4–14 CrossRef CAS PubMed.
- C. M. Micklitsch, S. H. Medina, T. Yucel, K. J. Nagy-Smith, D. J. Pochan and J. P. Schneider, Macromolecules, 2015, 48, 1281–1288 CrossRef CAS.
- A. Banerjee, G. Palui and A. Banerjee, Soft Matter, 2008, 4, 1430–1437 RSC.
- C. Tomasini and N. Castellucci, Chem. Soc. Rev., 2013, 42, 156–172 RSC.
- A. Aggeli, I. A. Nyrkova, M. Bell, R. Harding, L. Carrick, T. C. B. McLeish, A. N. Semenov and N. Boden, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 11857–11862 CrossRef CAS PubMed.
- V. Nebot, J. Armengol, J. Smets, S. Prieto, B. Escuder and J. Miravet, Chem.–Eur. J., 2012, 18, 4063–4072 CrossRef CAS PubMed.
- R. Duarte, R. Ongaratto, L. Piovesan, V. Lima, V. Soldi, A. Merlo and M. D'Oca, Tetrahedron Lett., 2012, 53, 2454–2460 CrossRef CAS.
- A. Ohta, K. Toda, Y. Morimota, T. Asakawa and S. Miyagishi, Colloids Surf., A, 2008, 317, 316–322 CrossRef CAS.
- S. Haldar and S. Maji, Colloids Surf., A, 2013, 420, 10–21 CrossRef CAS.
- J. Poolman, J. Boekhoven, A. Besselink, A. Olive, J. van Esch and R. Eelkema, Nat. Protoc., 2014, 9, 977–988 CrossRef CAS PubMed.
- R. Li, C. C. Horgan, B. Long, A. L. Rodriguez, L. Mather, C. J. Barrow, D. R. Nisbet and R. J. Williams, RSC Adv., 2015, 5, 301–307 RSC.
- L. Zhang, X. Wang, T. Wang and M. Liu, Small, 2015, 11(9–10), 1025–1038 CrossRef CAS PubMed.
- J. Bonnet, G. Suissa, M. Raynal and L. Bouteiller, Soft Matter, 2015, 11, 2308–2312 RSC.
- S. Haldar and K. Karmakar, RSC Adv., 2015, 5, 66339–66354 RSC.
- J. Merkel, J. Sturtevant and L. Regan, Structure, 1999, 7(11), 1333–1343 CrossRef CAS PubMed.
- S. Ahmed, J. Mondal, N. Behera and D. Das, Langmuir, 2013, 29, 14274–14283 CrossRef CAS PubMed.
- F. Menger and K. Caran, J. Am. Chem. Soc., 2000, 122, 11679–11691 CrossRef CAS.
- D. Jones and A. J. Middelberg, Langmuir, 2002, 18, 10357–10362 CrossRef CAS.
- T. V. Chalikian, High Pressure Bioscience: Basic Concepts, Application and Frontiers, ed. K. Akasak and H. Matsuki, Springer, 2015, ch. 3, p. 41 Search PubMed.
- F. Huang and W. M. Nau, Angew. Chem., 2003, 115, 2371–2374 CrossRef.
- R. Zhu and J. Dordick, Chem. Mater., 2006, 18, 5988–5995 CrossRef.
- J. Wu, T. Yi, Q. Xia, T. Shu, F. Liu, Y. Yang, F. Li, Z. Chen, Z. Zhou and C. Huang, J. Mater. Chem., 2009, 19, 3971–3978 RSC.
- G. Bellesia, M. Fedorov and E. Timoshenko, J. Chem. Phys., 2008, 128, 195105 CrossRef PubMed.
- D. de Jong, N. Lucas, M. Kellog, J. van Esch and L. Feringa, Science, 2004, 304, 278 CrossRef PubMed.
- K. Hanabusa, M. Yamada, M. Kimura and H. Shirai, Angew. Chem., Int. Ed., 1996, 35, 1949 CrossRef CAS.
- S. Bhattacharya and A. Pal, J. Phys. Chem. B, 2008, 112, 4918–4927 CrossRef CAS PubMed.
- S. Wheeler and K. N. Houk, J. Chem. Theory Comput., 2009, 5, 2301–2312 CrossRef CAS PubMed.
- S. Bromberg and K. Dill, Protein Sci., 1994, 3, 997–1009 CrossRef CAS PubMed.
- Y. Harpaz, M. Gerstein and C. Chothia, Structure, 1994, 2, 641 CrossRef CAS PubMed.
-
(a) T. V. Chalikian, High Pressure Bioscience: Basic Concepts, Application and Frontiers, ed. K. Akasak and H. Matsuki, Springer, 2015, ch. 3, p. 41 Search PubMed;
(b) M. Levitt, M. Gerstein, E. Huang, S. Subbiah and J. Tsai, Annu. Rev. Biochem., 1997, 66, 549–579 CrossRef CAS PubMed.
- J. Collier, B. Hu, J. Ruberti, J. Zhang, P. Shum, D. Thompson and P. Messersmith, J. Am. Chem. Soc., 2001, 123, 9463–9464 CrossRef CAS PubMed.
- T. Patra, A. Pal and J. Dey, Langmuir, 2010, 26(11), 7761–7767 CrossRef CAS PubMed.
-
(a) J. Eldridge and J. Ferry, J. Phys. Chem., 1954, 58, 992–995 CrossRef CAS;
(b) K. Kon-No, T. Jin-No and A. Kitahara, J. Colloid Interface Sci., 1974, 49, 383–389 CrossRef CAS;
(c) K. Murata, M. Aoki, T. Suzuki, T. Harada, H. Kawabata, T. Komori, F. Ohseto, K. Ueda and S. Shinkai, J. Am. Chem. Soc., 1994, 116, 6664–6676 CrossRef CAS;
(d) C. Garner, P. Terech, J. Allegraud, B. Mistrot, P. Nguyen, A. Geyer and D. Rivera, J. Chem. Soc., Faraday Trans., 1998, 94, 2173–2179 RSC;
(e) P. W. Atkins, Physical Chemistry, Oxford University Press, Oxford, U.K., 2nd edn, 1982, pp. 266–269 Search PubMed.
- D. Rizkov, J. Gun, O. Lev, R. Sicsic and A. Melman, Langmuir, 2005, 21, 12130–12138 CrossRef CAS PubMed.
- J. Makarević, M. Jokić, Z. Raza, Z. Štefanić, B. Kojić-Prodić and M. Žinić, Chem.–Eur. J., 2003, 9, 5567–5580 CrossRef PubMed.
- B. J. Stapley and J. Doig, J. Mol. Biol., 1997, 272, 456–464 CrossRef CAS PubMed.
- B. W. Chellgren and T. P. Creamer, Proteins: Struct., Funct., Bioinf., 2006, 62, 411–420 CrossRef CAS PubMed.
- P. Devidson, Molecular Gels: Materials with Self-Assembled Fibrillar Networks, ed. R. Weiss and P. Terech, Springer, 2006, ch. 26, p. 744 Search PubMed.
- F. Menger and A. Peresypkin, J. Am. Chem. Soc., 2003, 125, 5340–5345 CrossRef CAS PubMed.
- P. Terech, D. Pasquier, V. Bordas and C. Rossat, Langmuir, 2000, 16, 4485–4494 CrossRef CAS.
- R. L. Dunbrack and M. Karplus, Nat. Struct. Biol., 1994, 1(5), 334–340 CrossRef CAS PubMed.
- M. Laupheimer, N. Preisig and C. Stubenrauch, Colloids Surf., A, 2015, 469, 315–325 CrossRef CAS.
- M. Rogers, A. Wright and A. Marangoni, J. Phys. D: Appl. Phys., 2008, 41, 215501–215505 CrossRef.
- J. Wyman Jr, J. Am. Chem. Soc., 1931, 53, 3292–3301 CrossRef.
- E. Smith and P. Dea, Biophys. Chem., 2015, 196, 86–91 CrossRef CAS PubMed.
- Y. Deng, X. Feng, D. Yang, C. Yi and X. Qiu, BioResources, 2012, 7, 1145–1156 Search PubMed.
-
(a) W. Lee, J. Kim, C. Oh, T. Sakaguchi, M. Fujiki and G. Kwak, Angew. Chem., Int. Ed., 2010, 49, 1406–1409 CrossRef CAS PubMed;
(b) W. Z. Yuan, A. Qin, J. W. Y. Lam, J. Z. Sun, Y. Dong, M. Haussler, J. Liu, H. P. Xu, Q. Zhen and B. Z. Tang, Macromolecules, 2007, 40, 3159–3166 CrossRef CAS;
(c) A. Qin, C. K. W. Jim, Y. Tang, J. W. Y. Lam, J. Liu, F. Mahtab, P. Gao and B. Z. Tang, J. Phys. Chem. B, 2008, 112, 9281–9288 CrossRef CAS PubMed.
- T. Hinoue, Y. Shigenoi, M. Sugino, Y. Mizobe, I. Hisaki, M. Miyata and N. Tohnai, Chem.–Eur. J., 2012, 18, 4634–4643 CrossRef CAS PubMed.
- L. Bednárová, P. Maloň and P. Bouř, Chirality, 2007, 19, 775–786 CrossRef PubMed.
- J. Esch and F. Schoo, Langmuir, 2004, 20, 10851–10857 CrossRef PubMed.
- C. Wu, T. Sanborn, R. Zuckermann and A. Barron, JACS, 2001, 123, 2958–2963 CrossRef CAS PubMed.
- R. W. Woody, The Peptides: Analysis, Synthesis, Biology, ed. S. Udenfriend, J. Meienhofer and V. J. Hruby, Academic Press, 1985, vol. 7, ch. 2, p. 51 Search PubMed.
- C. Toniolo, Dynamic Aspect of Conformation changes in Biological Macromolecules, ed. C. Sadron, D. Reidel Publishing Co., Boston USA, 1973, p. 98 Search PubMed.
- V. Castelletto and I. W. Hamley, Biophys. Chem., 2009, 141, 169–174 CrossRef CAS PubMed.
- H. Dong, J. Mullol, J. Claret, L. Pérez, A. Pinazo and F. Sagués, Chem.–Eur. J., 2014, 20, 7396–7401 CrossRef CAS PubMed.
- A. Barth, Biochim. Biophys. Acta, Bioenerg., 2007, 1767, 1073–1101 CrossRef CAS PubMed.
- A. R. Hirst and D. K. Smith, Langmuir, 2004, 20, 10851–10857 CrossRef CAS PubMed.
- G. Zhu and J. Dordick, Chem. Mater., 2006, 18, 5988–5995 CrossRef CAS.
- K. Hanabusa, M. Matsumoto, M. Kimura, A. Kakehi and H. Shirai, J. Colloid Interface Sci., 2000, 224, 231–244 CrossRef CAS PubMed.
- A. Hirst, D. Smith, M. Feiters and H. Geurts, Langmuir, 2004, 20, 7070–7077 CrossRef CAS PubMed.
- J. Hardy, A. Hirst and D. Smith, Soft Matter, 2012, 8, 3399–3406 RSC.
- J. Makarević, M. Jokić, B. Perić, V. Tomišić, B. Kojić-Prodić and M. Žinić, Chem.–Eur. J., 2001, 7, 3328–3341 CrossRef.
- N. Zweep, A. Hopkinson, A. Meetsma, R. Browne, B. L. Feringa and J. H. van Esch, Langmuir, 2009, 25, 8802–8809 CrossRef CAS PubMed.
- Y. Loo, E. Wu, A. Lakshmanan, A. Mishra and C. Hauser, Self-Assembled Peptide Nanostructures: Advances and applications in nano biotechnology, ed. Jaime Castillo-León, Luigi Sasso and Winnie E. Svendsen, CRC Press, Taylor& Francis Group, 2012, ch. 6, p. 165 Search PubMed.
- A. Barton, Chem. Rev., 1975, 75, 731–753 CrossRef CAS.
- J. H. Hildebrand, Proc. Natl. Acad. Sci. U. S. A., 1979, 76(1), 194 CrossRef CAS.
- P. W. Snydera, J. Mecinovića, D. T. Moustakasa, S. W. Thomas, M. Hardera, E. T. Macka, M. R. Locketta, A. Hérouxb, W. Shermanc and G. M. Whitesidesa, Proc. Natl. Acad. Sci. U. S. A., 2011, 108(44), 17889–17894 CrossRef PubMed.
- T. Greaves, A. Weerawardena and C. Drummond, Phys. Chem. Chem. Phys., 2011, 13, 9180–9186 RSC.
- D. F. Evans, Langmuir, 1988, 4, 3–12 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra16797g |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.