
Differential in vitro and in vivo anti-angiogenic activities of acetal and ketal andrographolide derivatives in HUVEC and zebrafish models†
Abstract
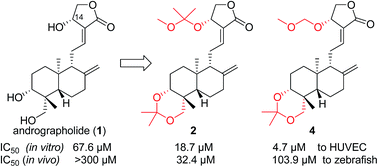
A series of acetal and ketal derivatives of andrographolide were synthesized and their anti-angiogenic activities were tested in vitro and in vivo using HUVEC and zebrafish models, respectively. These compounds exhibited better angiogenesis inhibitory activity in both models than the parent compound andrographolide (1). The compounds’ SARs differed for the HUVEC and zebrafish models, in that 14α-ketal 2 showed the best activity for in vivo anti-angiogenesis in zebrafish while 14α-acetals 4, 5 and 6 had greater in vitro anti-angiogenic activity with HUVECs than the other compounds and 1. The results suggested that methylene acetals 4, 5 and 6 were possibly hydrolyzed into 3 or 1 in zebrafish and that 14α-ketal 2 probably did not fully act as a pro-drug of 3 or 1 in zebrafish, instead exerting the anti-angiogenic effect itself or being metabolized into an unknown more active form(s) than 3 and 1 to block in vivo angiogenesis in zebrafish. The underlying molecular mechanisms of compound 2’s action were explored and the results indicated that VEGF-stimulated angiogenesis was significantly inhibited by compound 2 via targeting the phosphorylation of VEGFR2 and VEGFR2-mediated downstream angiogenesis signaling pathways. Therefore, this report demonstrates that andrographolide derivative(s) can be developed into therapeutic agent(s) against excessive angiogenesis, including tumor angiogenesis, after further improvement of the potency and stability of this series of andrographolide derivatives.
 Please wait while we load your content...
Please wait while we load your content...

