
Efficient and stereoselective synthesis of monomeric and bimetallic pincer complexes containing Pd-bonded stereogenic carbons†
Abstract
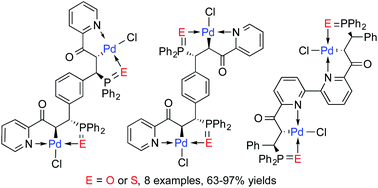
Enantiopure pyridyl-functionalized tertiary phosphines and diphosphines containing one or two stereogenic carbon centers were prepared efficiently by catalytic asymmetric hydrophosphination. The tertiary phosphines were oxidized to the corresponding oxides or sulfides by treatment with aq. H2O2 or elemental sulfur respectively. Chemoselective cyclopalladation of the phosphorus(V) species under mild reaction conditions generated the corresponding monomeric or bimetallic N–C(sp3)–E (E = O, S) Pd pincer complexes in high isolated yields (63–97%). All the new pincer complexes contain palladium-bound stereogenic carbons which are generated stereospecifically via the metal complexation process.
 Please wait while we load your content...
Please wait while we load your content...

