DOI:
10.1039/C6RA16713F
(Paper)
RSC Adv., 2016,
6, 85698-85703
Dihydropyridine-based fluorescence probe for nitric oxide†
Received
29th June 2016
, Accepted 1st September 2016
First published on 2nd September 2016
Abstract
A fluorescent probe for detecting nitric oxide has been designed based on dihydropyridine which acts as a switch-on unit. The optical properties of the probe feature a low background and high selectivity. Cellular assay revealed a significant response to nitric oxide produced in Raw 264.7 cells.
Introduction
Nitric oxide (NO) is a reactive free radical that is also known to be an atmospheric pollutant.1 NO plays important roles in a variety of physiological and pathological processes such as those found in cardiovascular, nervous, and immune systems.2 Regulations made by NO are related to various diseases including endothelial dysfunction, cancer and cardiovascular disease.3 In order to understand the mechanisms of NO, many techniques, such as electrochemistry,4 chemiluminescence,5 electron paramagnetic resonance,6 colorimetry,7 and fluorimetry have been developed.8 Due to its simplicity, high selectivity, sensitivity, and spatiotemporal resolution, fluorimetry has been considered as the most promising method for detecting NO.9
A large number of fluorescent probes based on small organic molecules or transition-metal complexes have been developed. For example, o-diaminobenzene derivatives first reported by Nagano's group,10 are well known and have already been applied to biological research. Metal complexes as fluorescent nitric oxide probes were reported by Lippard's group,11 which has provided a method for the direct detection of nitric oxide that has shown great potentials. However, probes based on o-diaminobenzene derivatives detected nitric oxide by operating under aerobic condition to generate the fluorescent triazole derivatives. Probes based on metal complexes for detecting nitric oxide were found to be sensitive to pH. As a result, fluorescent probes that are free of those limitations still need to be developed.
It is well known that 4-substituted derivatives of Hantzsch 1,4-dihydropyridine can be oxidized by nitric oxide (NO) to get the corresponding pyridine derivatives.12 Inspired by this reaction, we have designed a new strategy for detecting nitric oxide in living cells. Previously, we reported the synthesis and application of probe 1,13 which reacted with nitric oxide leading to fluorescence enhancement through oxidation of dihydropyridine. Although the compound we reported reacted with NO directly and selectively, the responds to NO was slow, which limited its application for NO detection in biological systems. To further improve the property of such kind of probes and to explore the structure–activity relationship, we reported here a new probe for NO detection, which demonstrated an unexpected high sensitivity and quick respond to nitric oxide. The basis of our strategy is shown in Scheme 1. The fluorescence of probe DHP-1 itself is very weak or non-fluorescent. Upon reacting with nitric oxide, the resultant product PY-1 turns strongly fluorescent. Probe DHP-1 consisted of a 7-methoxy coumarin as the fluorophore and dihydropyridine derivatives as the receptor for NO that are connected via a dimethylene linker.
 |
| | Scheme 1 Fluorescence turn-on for the probe DHP-1. | |
Results and discussions
Synthesis of probe DHP-1
The synthesis of DHP-1 is showed in Scheme 2. Intermediate 1a was synthesized by the condensation of 4-formyl-7-methoxycoumarin with malonic acid in pyridine. Compound 1a was hydrogenated with 10% palladium on activated carbon to get 1b. Compound 1c was prepared by reducing 1b with borane in tetrahydrofuran. Subsequent oxidation of 1c by Dess–Martin periodinane gave 1d. DHP-1 was formed by treating 1d with methyl acetoacetate in the presence of aqueous ammonia. Compound PY-1 was formed from the aromatization of DHP-1 by treating with NO. The disappearance of CH and NH protons on dihydropyridine ring indicated the aromatization of DHP-1, which was also supported by HR-MS. The identities of all the synthesized compounds were confirmed by NMR spectroscopy and mass spectrometry.
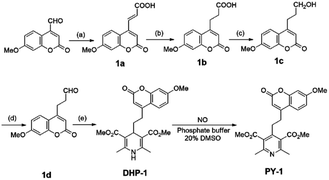 |
| | Scheme 2 Synthesis of probe DHP-1. Reagent and conditions: (a) malonic acid, pyridine, reflux; (b) Pd/C, H2, EtOH, 50 °C; (c) BH3·THF, 0 °C; (d) DMP, CH2Cl2, rt; (e) methyl acetoacetate, NH3·H2O, EtOH, reflux. | |
Fluorescence performances of probe DHP-1 toward NO
The optical property of DHP-1 was determined in a phosphate buffered solution (50 mM PBS, pH = 7.4, including 20% DMSO). DHP-1 exhibits a maximum linear absorption at 324 nm (ε = 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 530 M−1 cm−1) and a maximum emission at 392 nm, and is nearly non-fluorescent (ϕ < 0.01). Upon adding NO, the resultant PY-1 exhibits a maximum linear absorption at 323 nm (ε = 84900 M−1 cm−1) and a maximum emission at 392 nm, with although no shift in the maximum absorption and emission wavelengths being detected. However, the fluorescence intensity of PY-1 showed a dramatic enhancement of about 80-fold at 392 nm (ϕ = 0.91) over that of DHP-1 (Fig. 1), which showed higher enhancement and quantum yield than those of the probe we reported previously (40-fold enhancements and ϕ = 0.39). Such high enhancement is really unexpected considering the saturated dimethylene bridge between fluorophore and aromatized pyridine units, which might attribute to the efficient overlap of the HOMO and LUMO for PY-1.
530 M−1 cm−1) and a maximum emission at 392 nm, and is nearly non-fluorescent (ϕ < 0.01). Upon adding NO, the resultant PY-1 exhibits a maximum linear absorption at 323 nm (ε = 84900 M−1 cm−1) and a maximum emission at 392 nm, with although no shift in the maximum absorption and emission wavelengths being detected. However, the fluorescence intensity of PY-1 showed a dramatic enhancement of about 80-fold at 392 nm (ϕ = 0.91) over that of DHP-1 (Fig. 1), which showed higher enhancement and quantum yield than those of the probe we reported previously (40-fold enhancements and ϕ = 0.39). Such high enhancement is really unexpected considering the saturated dimethylene bridge between fluorophore and aromatized pyridine units, which might attribute to the efficient overlap of the HOMO and LUMO for PY-1.
 |
| | Fig. 1 UV-vis absorption and fluorescence emission spectra of probe DHP-1 (black) and compound PY-1 (red) (1 μM in phosphate buffer including 20% DMSO, 50 mM, pH 7.4 for both DHP-1 and PY-1). | |
Detecting mechanism of probe DHP-1 toward NO
To gain insights into the observed difference in the fluorescent intensities of DHP-1 and PY-1, quantum chemical calculations were carried out using the Gaussian 09 program package.14 Their energy levels of frontier molecular orbitals have been optimized with density functional theory (DFT) at the B3LYP/6-31+G(d) level,15 and are shown in Fig. 2 and 3. It can be seen that the electronic wave function overlap between HOMO and LUMO of the fluorophore on DHP-1 is obviously insufficient and thus the transition is inefficient or “forbidden”. For PY-1, the HOMO and LUMO on coumarin moiety is well overlapped, and results in efficient electronic transition with high fluorescent quantum yield. Moreover, the energy of the HOMO for DHP-1 is −5.72 eV which is almost isoenergetic with that of HOMO−1 (−5.79 eV). However, for compound PY-1, the energy of HOMO−1 (−6.57 eV) is much lower than HOMO (−5.91 eV). This decrease of HOMO−1 level greatly disfavors the crossing from the emissive locally excited state to the dark electron transfer state, thus prevents PET quench mechanism. The result of DFT calculation clearly confirms that the fluorescence enhancement of PY-1 results from the suppression of nonradiative pathway after the NO-induced aromatization. Therefore, the probe DHP-1 is a sensor with an “off–on” property for nitric oxide detection.16
 |
| | Fig. 2 The key molecular orbitals for DHP-1 and PY-1. | |
 |
| | Fig. 3 Relative energies of the molecular orbitals for the ground states of DHP-1 (left) and PY-1 (right). The HOMO−1 of PY-1 is too low to quench fluorescence. | |
pH dependence of compound PY-1
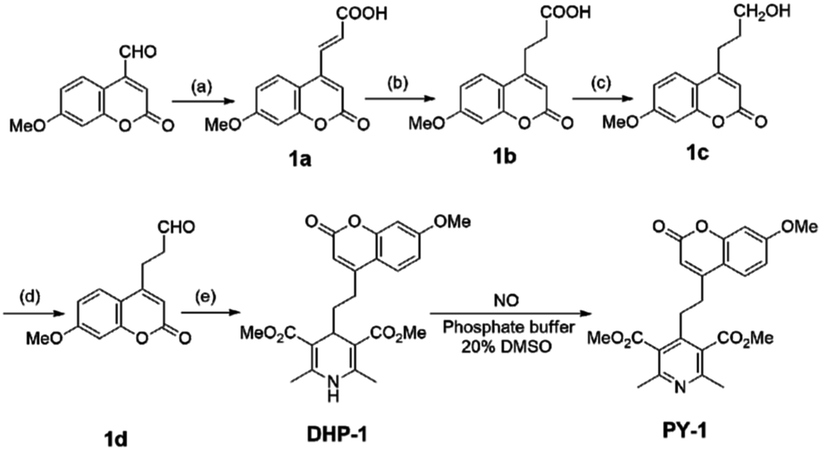
As pH independent fluorescence sensing is very important for biological applications, the effect of pH on fluorescence intensity upon treating DHP-1 with NO was tested (Fig. 4). The reaction product of DHP-1 with NO exhibits stable fluorescence intensity in the pH range from 4 to 9, which indicates that the fluorescence intensity of compound PY-1 remains unchanged over a wide pH range.
 |
| | Fig. 4 The pH dependence of the fluorescence intensity of PY-1 (2.5 μM) in phosphate buffer solution with 20% DMSO. λex = 323 nm, λem = 392 nm. | |
Reaction kinetics of probe DHP-1 toward NO
Moreover, the reaction for DHP-1 with NO was so fast that obvious fluorescence enhancement was observed in about 2 min after adding 1.2 equivalents of NO (Fig. 5), the maximum change was reached after 10 min, while it took about 90 min for our previous probe at the same condition. The rate constant of the reaction was calculated by fitting the curve of the fluorescent intensity versus time using pseudo-first-order equation, which is 0.31 min−1.17 The fast NO response of DHP-1 will benefit its application in biological systems.
 |
| | Fig. 5 Fluorescence spectra changing for the reaction of DHP-1 (2.5 μM) with 1.2 equiv. of NO-saturated solution over the time (0, 1, 2, 5, 10, 15, 20 min) (λex = 323 nm, λem = 392 nm) under air. | |
Sensitivity and selectivity of probe DHP-1 for NO
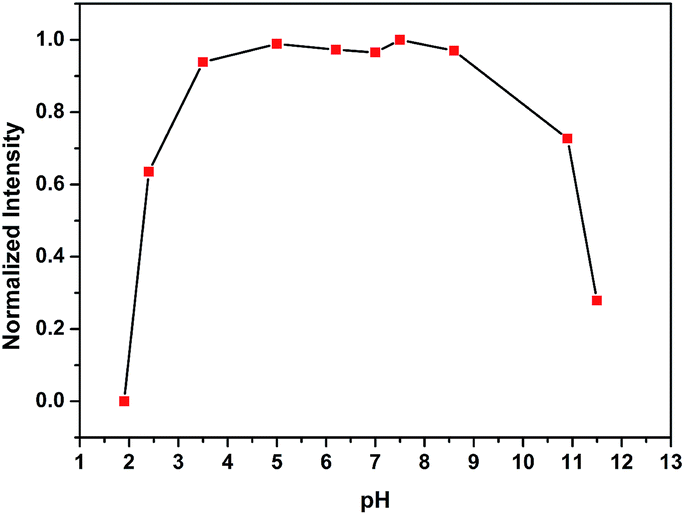
The fluorescence spectra for the reaction of probe DHP-1 with different equivalent of NO-saturated solution for 10 min were detected (Fig. 6). Upon the addition of different equivalent of NO-saturated solution, significant change in fluorescence was detected, which indicates that probe DHP-1 can response to the concentration of NO. The detection limit of probe DHP-1 was calculated to be 17.0 nM.18
 |
| | Fig. 6 Fluorescence spectra upon addition of NO saturated solution from 0–1.0 equiv. to a solution of DHP-1 (2.5 μM) in 50 mM sodium phosphate buffer at pH 7.4 including 20% DMSO (λex = 323 nm) under air. | |
The responses of DHP-1 to NO and other reactive nitrogen or oxygen oxides that exist in living organisms were compared (Fig. 7). Except for NO, the presence of NO2−, NO3−, H2O2, OONO−, ClO−, ascorbic acid (AA), O2˙− or 1O2, failed to cause significant enhancement in fluorescence intensity above background (blank). Those results indicate that probe DHP-1 has high selectivity for NO.
 |
| | Fig. 7 Fluorescence intensity of DHP-1 (2.5 μM, in phosphate buffer including 20% DMSO, 50 mM, pH 7.4) at 392 nm upon the addition of 100 equivalents of various reactive species: NO2−, NO3−, H2O2, OONO−, NO, ClO−, ascorbic acid (AA), O2˙−, 1O2, λex = 323 nm. | |
Imaging NO in living cells
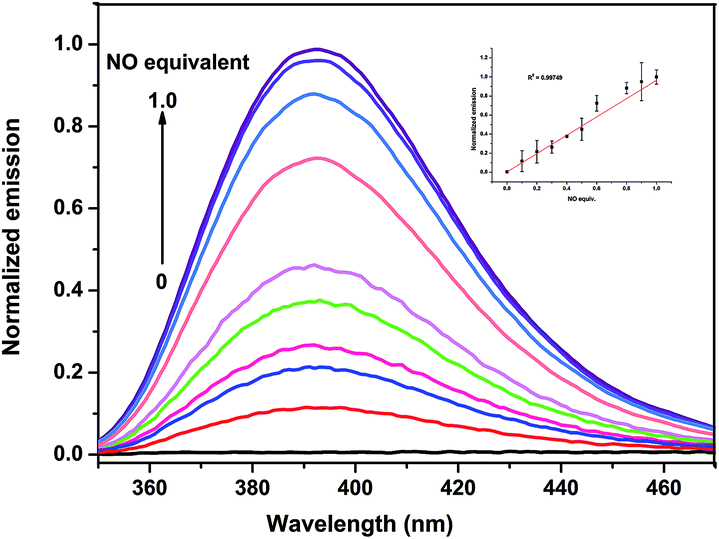
In order to evaluate the efficacy of DHP-1 in sensing endogenously produced NO, Raw 264.7 macrophage cells, which were often used in the NO detection,19 incubated with probe DHP-1 in the absence of bacterial lipopolysaccharide (LPS) display very weak fluorescence (Fig. 8a). When the Raw 264.7 macrophage cells were pretreated with bacterial lipopolysaccharide (LPS) which promotes NO production by inducible nitric oxide synthase (iNOS),20 significant fluorescence enhancement is observed (Fig. 8b). This result shows that probe DHP-1 can detect LPS-stimulated endogenous NO produced in living cells. Moreover, when stimulated cells were pretreated with NG-methyl-L-arginine (L-NMA),21 a selective inhibitor of LPS-stimulated NO production (Fig. 8c), the fluorescence was found to be similar to that of untreated cells. This result indicates that the significant fluorescence in the LPS-stimulated cells is indeed due to the direct reaction of probe DHP-1 with NO.
 |
| | Fig. 8 Probe DHP-1 detection of endogenous NO in Raw 264.7 cells: bright field image (top) and corresponding fluorescence image (bottom) of cells (a) incubated with probe DHP-1 (5 μM) without LPS prestimulation, (b) prestimulated with LPS (0.5 μg mL−1) for 4 h and incubated with probe DHP-1 (5 μM) for 8 h, the fluorescence intensity is 0.016 ± 0.0012 (n = 3), (c) pretreated with NOS inhibitor (L-NMA, 2 mM) 1.5 h and incubated with LPS 4 h then probe DHP-1 (5 μM) 8 h. Scale bars are all 100 μm. Emission was collected at 445 ± 50 nm (λex = 365 nm). | |
Cytotoxicities of DHP-1 and PY-1
The cytotoxicities of DHP-1 and PY-1 were examined by MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) method. The obtained results showed that over 80% of Raw 264.7 cells survived after incubation with probe DHP-1 or PY-1 at different concentrations, which suggests that neither DHP-1 nor compound PY-1 is toxic to Raw 264.7 cells in the concentration range adopted for the imaging studies (Fig. 9).
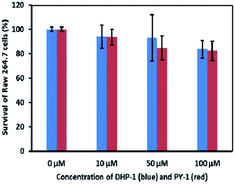 |
| | Fig. 9 MTT assay of Raw 264.7 cells treated with DHP-1 and PY-1. | |
Experimental
Materials and methods
Solvents and reagents were purchased from commercial suppliers and used without further purification unless otherwise indicated. Spectroscopic grade DMSO were used for spectroscopic studies. All NMR spectra were obtained on Bruker 400 MHz spectrometer. Chemical shifts were reported in parts per million (ppm) downfield from TMS (tetramethylsilane). Mass spectra were obtained from a Quattro microtriple quadrupole mass spectrometer (Waters, Milford, MA, USA). UV-vis spectra were recorded on a Cary-50 UV-vis spectrophotometer. Fluorescence spectra were recorded on a Cary Eclipse Fluorescence spectrophotometer (V = 600 volt). The adjustable slit was set at 5 nm. Fluorescence quantum yield of samples were recorded on a Fluoromax-4 spectrophotometer at room temperature with an integrating sphere system, and the machine was revaluated using standard sample before measurement. Melting points were taken on a X-5B precise micro melting point apparatus. FT-IR spectra were taken on an IR Affinity-1 Fourier Transform Infrared Spectrometer in the range of 4000–400 cm−1 (Thermo Fisher Nicolet, USA). All pH measurements was determined by Five Easy Plus FEP20 pH meter (Mettler-Toledo Instruments (Shanghai) Co., LTD.). The nitric oxide (NO) stock solution in de-ionized water was prepared according to the literature.22 Peroxynitrite was generated from amyl nitrite and H2O2.23 Quartz cuvettes with 10 mm path lengths and four faces polished were used. Stock solutions of probe DHP-1 (5 mM) were prepared in phosphate buffer (50 mM, pH 7.4) with 20% DMSO.
Cell culture and imaging materials
Raw 264.7 cells were obtained from Cell Resource Center (IBMS, CAMS/PUMC) and cultured in Dulbecco's Modified Eagle Medium (DMEM; Thermo Scientific) supplemented with 10% fetal bovine serum (FBS; GIBCO; Invitrogen), and 1% penicillin/streptomycin (Beijing Solarbio Scientific & Technology Co, Ltd). For imaging studies, cells were plated in Class Bottom Cell Culture Dish (Nest) containing 1 mL of complete DMEM and incubated at 37 °C under 5% CO2 for one day. To induce NO production, Raw 264.7 cells were stimulated with 0.5 μg mL−1 lipopolysaccharide (LPS, Sigma-Aldrich) for 4 h. Bright field and fluorescence images were taken with a Zeiss Observer A1 inverted fluorescence microscope equipped with an EM-CCD camera (Hamamatsu) and an X-Cite 120 metal halide lamp (EXFP). Bright field image and fluorescence images were obtained using an 40× objective lens.
Cyotoxicity assays
Raw 264.7 cells were grown in 96-well plates at an initial density 1 × 103 cells per well for 24 h. Subsequently, the 10 μM, 50 μM and 100 μM of probe DHP-1 and compound PY-1 were incubated for 12 h, respectively. Cell viability was evaluated using the 3-(4,5-dimethylthiazol-2-yl)2,5-diphenyl-tetrazolium bromide (MTT) reduction assay. After incubation, MTT (20 μL, 5 mg mL−1) was added to each well for 4 h, then DMSO (100 μL) was added to each well after removing media. Absorption at 490 nm was measured on a plate reader.
Experiments and characterizations
The synthesis of 1a. A mixture of 4-formyl-7-methoxy cumarin (4.00 g, 19 mmol), malonic acid (6.12 g, 29.4 mmol) and pyridine (10 mL) was refluxed for 1 h. The reaction was poured into water and then acidified with concentrated HCl, then the suspension was filtered, and the precipitate was recrystallized by ethanol to afford yellow solid (2.24 g, yield 48%). Mp: 230–232 °C, 1H NMR (400 MHz, DMSO-d6) δ 7.86 (d, J = 15.8 Hz, 1H), 7.81 (d, J = 8.9 Hz, 1H), 7.05 (d, J = 2.5 Hz, 1H), 6.99 (dd, J = 2.5, 2.5 Hz, 1H), 6.76 (d, J = 15.8 Hz, 1H), 6.63 (s, 1H), 3.87 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 166.3, 162.6, 159.9, 155.1, 147.5, 135.8, 128.2, 126.1, 112.3, 110.9, 109.6, 101.1, 55.9. ESI MS calculated 246.0, found 245.3 [M − H]−. IR (KBr) ν 3070, 2984, 2665, 1723, 1688, 1607, 1547, 1379, 1287, 1143, 988, 971, 863, 820.
The synthesis of 1b. A mixture of 1a (520 mg, 2.11 mmol) and 5% Pd/C (104 mg) in ethanol (50 mL) was hydrogenated at an initial H2 pressure, and the mixture was heated to 50 °C. 6 h later, the reaction mixture was filtered through a Celite, and the filtrate was removed to obtain oil. The oil was dissolved in 5% sodium bicarbonate, the aqueous solution was filtered and the filtrate was acidified with concentrated HCl, then filtered, white solid was obtained (400 mg, yield 76%). Mp: 177–179 °C, 1H NMR (400 MHz, DMSO-d6) δ 7.76 (d, J = 8.8 Hz, 1H), 7.01 (d, J = 2.3 Hz, 1H), 6.98 (dd, J = 2.4, 2.4 Hz, 1H), 6.15 (s, 1H), 3.86 (s, 3H), 3.03 (t, J = 7.3 Hz, 2H), 2.66 (t, J = 7.6 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 177.1, 162.2, 160.1, 155.3, 154.8, 125.9, 112.1, 109.9, 100.9, 55.8, 31.7, 26.0. ESI MS calculated 248.1, found 249.2 [M + H]+. IR (KBr) ν 3068, 2844, 1735, 1588, 1516, 1393, 1213, 1137, 1023, 985, 874, 740.
The synthesis of 1c. A solution of 1b (100 mg, 0.41 mmol) in dry methylene chloride (5 mL) was cooled to 0 °C under ice bath, a 1.0 M solution of borane in THF (3.25 mmol, 3.2 mL) was added dropwisely. The mixture was reached to room temperature and stirred for 1.5 h. The mixture was cooled to 0 °C again, and methanol was added slowly. The mixture was evaporated under vacuum and the residue was dissolved in ethyl acetate, washed with water, dried over anhydrous sodium sulfate, filtered. The filtrate was evaporated and the crude product was purified by chromatography (chloroform/methanol = 20:1) to obtained white solid (60 mg, yield 63%). Mp: 126–128 °C, 1H NMR (400 MHz, CDCl3) δ 7.58 (d, J = 8.7 Hz, 1H), 6.87 (d, J = 2.5 Hz, 1H), 6.85 (dd, J = 2.5, 2.5 Hz, 1H), 6.16 (s, 1H), 3.87 (s, 3H), 3.80 (t, J = 6.0 Hz, 2H), 2.88 (t, J = 7.0 Hz, 2H), 1.98–1.91 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 162.6, 161.5, 156.0, 155.5, 125.3, 112.7, 112.3, 110.9, 101.0, 61.6, 55.8, 30.9, 28.0. ESI MS calculated 234.1, found 234.5 [M + H]+. IR (KBr) ν 3490, 3082, 2945, 2853, 1708, 1607, 1403, 1210, 1136, 1022, 986, 839.
The synthesis of 1d. To a solution of 1c (50 mg, 0.22 mmol) in dry methylene chloride (5 mL) was added Dess–Martin oxidation (182 mg, 0.43 mmol), the mixture was stirred at room temperature for 30 min. Saturated sodium bicarbonate was added to quench the reaction, then collect organic layer, dried over anhydrous sodium sulfate, filtered. The filtrate was evaporated and the crude product was purified by chromatography (ethyl acetate/petroleum ether = 1:2) to obtain light yellow solid (22 mg, yield 44%). Mp: 102–104 °C, 1H NMR (400 MHz, CDCl3) δ 9.89 (s, 1H), 7.52 (d, J = 8.8 Hz, 1H), 6.88 (dd, J = 2.5, 2.5 Hz, 1H), 6.83 (d, J = 2.3 Hz, 1H), 6.11 (s, 1H), 3.87 (s, 3H), 3.09 (t, J = 7.0 Hz, 2H), 2.92 (t, J = 7.6 Hz, 2H). 13C NMR (101 MHz, CDCl3) δ 199.2, 162.8, 161.0, 155.5, 154.2, 124.9, 112.5, 110.9, 101.2, 55.8, 41.6, 23.6. ESI MS calculated 232.1, found 231.4 [M − H]−. IR (KBr) ν 3066, 2915, 2838, 1750, 1704, 1610, 1397, 1300, 1147, 1039, 874, 833, 745.
The synthesis of DHP-1. 1d (300 mg, 1.29 mmol), methyl acetoacetate (300 mg, 2.59 mmol) and ammonia solution (91 mg, 2.59 mmol) were dissolved in 5 mL ethanol and refluxed for 1.5 hours. After cooling to room temperature, the reaction mixture was concentrated in vacuo to give crude product which was purified by chromatography (ethyl acetate/petroleum ether = 1:1) to afford DHP-1 (65 mg, yield 12%) as white solid. Mp: 204–206 °C, 1H NMR (400 MHz, CDCl3) δ 7.42 (d, J = 8.6 Hz, 1H), 6.83 (d, J = 2.5 Hz, 1H), 6.81 (dd, J = 2.5, 2.5 Hz, 2H), 6.13 (s, 1H), 5.84 (s, 1H), 4.11–4.07 (m, 1H), 3.86 (s, 3H), 3.71 (s, 6H), 2.76–2.63 (m, 2H), 2.34 (s, 6H), 1.72–1.66 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 167.3, 162.0, 160.1, 156.6, 154.9, 147.0, 125.3, 112.2, 109.6, 100.9, 100.0, 55.8, 50.5, 34.3, 32.0, 26.2, 18.1. HR-MS calculated 450.1529, found 450.1522 [M + Na]+. IR (KBr) ν 3346, 2956, 2931, 1730, 1707, 1647, 1483, 1431, 1282, 1220, 839, 783.
The synthesis of PY-1. NO gas was bubbled through a CH2Cl2 solution containing DHP-1 (100 mg, 0.234 mmol) at room temperature and the reaction was monitored by TLC. The reaction mixture was sequentially washed with saturated NaHCO3 and NaCl solutions, and dried over anhydrous MgSO4 and evaporated in vacuo. The resulting residue as crude product was purified by flash chromatography with a mixture ethyl acetate and petroleum ether (1:1) as eluent to afford PY-1 as yellowish crystalline (48 mg, yield 48%). Mp: 147–149 °C, 1H NMR (400 MHz, CDCl3) δ 7.61 (d, J = 8.8 Hz, 1H), 6.89 (dd, J = 2.5, 2.5 Hz, 1H), 6.84 (d, J = 2.4 Hz, 1H), 6.10 (s, 1H), 3.91 (s, 3H), 3.88 (s, 6H), 3.01–2.89 (m, 2H), 2.88–2.85 (m, 2H), 2.54 (s, 6H). 13C NMR (101 MHz, DMSO-d6) δ 167.9, 162.4, 159.9, 155.2, 154.4, 144.0, 126.5, 125.5, 112.1, 110.8, 101.1, 55.9, 52.7, 31.8, 29.9, 22.6. HR-MS calculated 426.1553, found 426.1551 [M + H]+. IR (KBr) ν 2953, 2924, 2852, 1714, 1608, 1560, 1396, 1247, 1035, 844, 792.
Conclusions
In conclusion, we have developed the rationally designed, synthetically readily available DHP-1 for detecting NO. The optical characteristics of and living cell imaging with probe DHP-1, revealed a rapid response to NO with 80-fold fluorescence intensity enhancement compared with background, and a high selectivity for NO over other reactive oxygen or nitrogen species. Probe DHP-1 is also applicable for NO detection in living cells at biologically relevant pH range.
Acknowledgements
The authors gratefully acknowledge the support from the National Natural Science Foundation of China (21272274), the Training Program of the Major Research Plan of the NSFC Grant (91227109), the National S&T Major Special Project on Major New Drug Innovation Grant (2015ZX09303-001), the Special Fund for Agro-Scientific Research in the Public Interest Grant (201103027).
Notes and references
- R. F. Furchgott, Angew. Chem., Int. Ed., 1999, 38, 1870–1880 CrossRef CAS.
-
(a) S. Moncada, R. M. J. Palmer and E. A. Higgs, Pharmacol. Rev., 1991, 43, 109–142 CAS;
(b) R. Butlerand and D. L. Williams, Chem. Soc. Rev., 1993, 22, 233–241 RSC;
(c) L. Jia, C. Bonaventura, J. Bonaventura and J. S. Stamler, Nature, 1996, 380, 221–226 CrossRef CAS PubMed.
-
(a) C. S. Cobbs, J. E. Brenman, K. D. Aldape, D. S. Bredt and M. A. Israel, Cancer Res., 1995, 55, 727–730 CAS;
(b) G. P. Biro, Curr. Drug Discovery Technol., 2012, 9, 194–203 CrossRef CAS;
(c) R. J. Giedt, C. J. Yang, J. L. Zweier, A. Matzavinos and B. R. Alevriadou, Free Radical Biol. Med., 2012, 52, 348–356 CrossRef CAS PubMed;
(d) D. Y. Choi, Y. J. Lee, J. T. Hong and H. J. Lee, Brain Res. Bull., 2012, 87, 144–153 CrossRef CAS PubMed.
- F. Bediouiand and N. Villeneuve, Electroanalysis, 2003, 15, 5–18 CrossRef.
-
(a) J. F. Brien, B. E. Mclaughlin, K. Nakatsu and G. S. Marks, Methods Enzymol., 1996, 268, 83–92 CAS;
(b) S. S. Cui, H. Y. Chen, Y. Z. Hu and Y. Q. Gu, Chin. Pharm. J., 2010, 45, 1524–1527 CAS.
- T. Naganoand and T. Yoshimura, Chem. Rev., 2002, 102, 1235–1270 CrossRef.
- L. A. Ridnour, J. E. Sim, M. A. Hayward, D. A. Wink, S. M. Martin, G. R. Buettner and D. R. Spitz, Anal. Biochem., 2000, 281, 223–229 CrossRef CAS PubMed.
- H. Kojima, N. Nakatsubo, K. Kikuchi, S. Kawahara, Y. Kirino, H. Nagoshi, Y. Hirata and T. Nagano, Anal. Chem., 1998, 70, 2446–2453 CrossRef CAS PubMed.
- S. A. Hilderbrand, M. H. Lim and S. J. Lippard, Topics in Fluorescence Spectroscopy, 2005, vol. 9, pp. 163–188 Search PubMed.
-
(a) T. Nagano and T. Yoshimura, Chem. Res., 2002, 102, 1235–1269 CAS;
(b) T. P. Misko, R. J. Schilling, D. Salvemini, W. M. Moore and M. G. Currie, Anal. Biochem., 1993, 214, 11–16 CrossRef CAS PubMed;
(c) P. G. Gunasekar, A. G. Kanthasamy, J. L. Borowitz and G. E. Isom, J. Neurosci. Methods, 1995, 61, 15–21 CrossRef CAS PubMed;
(d) A. Imrich and L. Kobzik, Nitric Oxide, 1997, 1, 359–369 CrossRef CAS PubMed;
(e) H. Kojima, Y. Urano, K. Kikuchi, T. Higuchi, Y. Hirata and T. Nagano, Angew. Chem., Int. Ed., 1999, 38, 3209–3210 CrossRef CAS.
-
(a) M. H. Lim and S. J. Lippard, J. Am. Chem. Soc., 2005, 127, 12170–12171 CrossRef CAS PubMed;
(b) M. H. Lim, D. Xu and S. J. Lippard, Nat. Chem. Biol., 2006, 2, 375–380 CrossRef CAS PubMed;
(c) R. C. Smith, A. G. Tennyson, M. H. Lim and S. J. Lippard, Org. Lett., 2005, 7, 3573–3575 CrossRef CAS PubMed;
(d) L. E. McQuade, J. Ma, G. Lowe, A. Ghatpande, A. Gelperin and S. J. Lippard, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 8525–8530 CrossRef CAS PubMed;
(e) M. D. Pluth, M. R. Chan, L. E. McQuade and S. J. Lippard, Inorg. Chem., 2011, 50, 9385–9392 CrossRef CAS PubMed.
- X. Q. Zhu, B. J. Zhao and J. P. Cheng, J. Org. Chem., 2000, 65, 8158–8163 CrossRef CAS PubMed.
- S. F. Ma, D. C. Fang, B. M. Ning, M. F. Li, L. He and B. Gong, Chem. Commun., 2014, 50, 6475–6478 RSC.
- M. J. Frisch, G. W. Trucks and H. B. Schlegel, et al., Gaussian 09, Gaussian, Inc., Wallingford CT, 2010 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- M. H. Lim, B. A. Wong and S. J. Lippard, J. Am. Chem. Soc., 2006, 128, 14364–14373 CrossRef CAS PubMed.
-
(a) P. B. Pati and S. S. Zade, RSC Adv., 2013, 3, 13457–13462 RSC;
(b) X. Y. Sun, Y. Wang, X. F. Zhang, S. H. Zhang and Z. Zhang, RSC Adv., 2015, 5, 96905–96910 RSC.
- B. C. Zhu, C. C. Gao, Y. Z. Zhao, C. Y. Liu, Y. M. Li, Q. Wei, Z. M. Ma, B. Du and X. L. Zhang, Chem. Commun., 2011, 47, 8656–8658 RSC.
-
(a) C. C. Chen and J. K. Wang, Mol. Pharmacol., 1999, 55, 481–488 CAS;
(b) G. K. Vegesna, S. R. Sripathi, J. T. Zhang, S. L. Zhu, W. L. He, F. T. Luo, W. J. Jahng, M. Ftost and H. Y. Liu, ACS Appl. Mater. Interfaces, 2013, 5, 4107–4112 Search PubMed;
(c) X. L. Sun, G. Kim, X. F. Xu, J. Y. Yoon and T. D. James, ChemPlusChem, 2016, 81, 30–34 CrossRef CAS;
(d) M. H. Lim, D. Xu and S. J. Lippard, Nat. Chem. Biol., 2006, 2, 375–380 CrossRef CAS PubMed.
- C. Bogdan, Nat. Immunol., 2001, 2, 907–916 CrossRef CAS PubMed.
-
(a) R. G. Kibourn, A. Jrbran, S. S. Gross, O. W. Griffith, R. Levi, J. Adams and R. F. Lodato, Biochem. Biophys. Res. Commun., 1990, 172, 1132–1138 CrossRef;
(b) A. Beltrán, M. I. Burguete, D. R. Abánades, D. Pérez-Sala, S. V. Luis and F. Galindo, Chem. Commun., 2014, 50, 3579–3581 RSC.
- H. Yu, X. Zhang, Y. Xiao, W. Zou, L. Wang and L. Jin, Anal. Chem., 2013, 85, 7076–7084 CrossRef CAS PubMed.
- M. Uppu and W. A. Pryor, Anal. Biochem., 1996, 236, 242–249 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra16713f |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.