
Environmentally sensitive nanohydrogels decorated with a three-strand oligonucleotide helix for controlled loading and prolonged release of intercalators†
Wioletta Liwinskaa,
Michał Symonowicza,
Iwona Stanislawskab,
Marek Lypb,
Zbigniew Stojeka and
Ewelina Zabost*a
aFaculty of Chemistry, University of Warsaw, ul. Pasteura 1, 02-093 Warsaw, Poland. E-mail: ezabost@chem.uw.edu.pl
bCollege of Rehabilitation, Kasprzaka 49, Warsaw, Poland
First published on 16th September 2016
Abstract
Two different short DNA strands were attached to the surface of a gel nanoparticle. The third DNA strand that was 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 complementary to those strands allowed the formation of a three-strand hybrid. The gel nanoparticles were synthetized from N-isopropyloacrylamide (NIPA), N,N′-methylenebisacrylamide (BIS) and acrylic acid (AA) by employing the free radical polymerization reaction. The physicochemical parameters of this novel nanogel stimulated its penetration of selected cancer tissues (Hela and Insulinoma cells) and allowed effective delivery of the anticancer drug doxorubicin (Dox). Since the three-strand oligonucleotide hybrid sites were located at the surface of the nanogel, this allowed effective storing of Dox by its intercalation to the double stranded DNA. The binding through intercalation resulted in prolonged release of the drug. The release of Dox at selected temperatures was a consequence of oligo1-2-3 hybrid conformational change, the shrinking of the hydrogel and zeta-potential change.
:50 complementary to those strands allowed the formation of a three-strand hybrid. The gel nanoparticles were synthetized from N-isopropyloacrylamide (NIPA), N,N′-methylenebisacrylamide (BIS) and acrylic acid (AA) by employing the free radical polymerization reaction. The physicochemical parameters of this novel nanogel stimulated its penetration of selected cancer tissues (Hela and Insulinoma cells) and allowed effective delivery of the anticancer drug doxorubicin (Dox). Since the three-strand oligonucleotide hybrid sites were located at the surface of the nanogel, this allowed effective storing of Dox by its intercalation to the double stranded DNA. The binding through intercalation resulted in prolonged release of the drug. The release of Dox at selected temperatures was a consequence of oligo1-2-3 hybrid conformational change, the shrinking of the hydrogel and zeta-potential change.
1. Introduction
In recent years, due to the rapid development of nanotechnology, various nanostructured multicomponent drug delivery systems (DDSs) have been proposed. Polymeric-based nanosystems which included liposomes, nanoparticles, micelles, nanolayers and nanogels in combination with metallic- and semiconductor nanoparticles and biomolecules have often been employed.1–4 Increased effectiveness of drug performance and in general positive results were reported.5Polymeric nanogels became attractive due to their potential application in the field of biomedical technologies. They possess cross-linked 3D networks that offer a load of the solution of up to 95% and environmental sensitivity to various external stimuli.6,7 Their stimulus responsiveness is a result of their ability to undergo the volume phase transition or the swelling–shrinking process in response to changes in external physicochemical parameters such as pH, temperature, redox potential, ionic strength, solvent composition and presence of various small molecules.8–10 Nanogels that are useful as DDSs often involve prolonged action time, enhanced drug protection, improved accumulation of drugs and enhanced delivery to cancer tissues thanks to increased permeability and reduced adverse effects.11–13 Typical ways of drug loading into nanogel nets are: equilibrium partitioning between the solution and the nanogels phases, electrostatic- and hydrophobic interactions and hydrogen- and biodegradable covalent bonding.14–17 The introduction, into a hydrogel interior network, of biological components such as oligonucleotides, offer an additional option of loading of drugs into nanogel nets: this is by intercalation of the drug between the parallel planes of nucleic bases of double stranded DNA.18–20
Hybridization of DNA strands was also employed in the construction of macro- and micro-sized hydrogel networks. Generally, two strategies were applied: (a) design of spatial networks with either linear or branched DNA-based copolymers and (b) grafting of linear polymers by oligonucleotide strands.21–23 The application of DNA hybridization in the construction of DNA-based shells around several types of solid nanoparticles was also reported.24–26 Another approach was the covalent grafting of aptamers onto solid- and polyelectrolyte types of microcapsules and linear polymeric hydrogels, where the single and duplex species could be separated and could specifically interact with drug molecules.22–29 Aptamers are short oligonucleotide strands specifically sequentially self-organized that can recognize low-weight molecules, biomolecules and cells overexpressed in tumor tissues. Explicitly, the formation of aptamer–ligand complexes may trigger a stimuli-responsiveness of hydrogels.24,28
The presence of double stranded DNA and its ability to undergo reversible conformational changes in response to temperature, gives a potential for reversible modulation of phase transition of the hydrogel lattices and the achievement of selectiveness of drug binding and prolonged release of drugs.18,19 The influence of ionic strength and temperature on stability of multilayers of oligonucleotides of different length was also presented.30 To our best knowledge, an analysis of the correlation between structural changes of hybridized DNA, interlinked in thermoresponsive nanogel nets, and improved storing and prolonged release of intercalators was not reported yet.
In the present work, we propose a novel drug carrier that is based on copolymer PNIPA-co-AAc nanohydrogel. AAc copolymers were already applied for the release of selected drugs at elevated temperature.31,32 The novelty is a specifically sequentially programmed three-strand hybridized oligonucleotide system built into the surface of the nanogels. PNIPA-co-AAc-based nanogels were first covalently decorated with two types of Acrydite®-modified oligonucleotide strands: oligo1 and oligo2. Then oligo3 that is 50% complementary to both: oligo1 and oligo2 strands was attached by hybridization with oligo1 and oligo2 to the nanogels. The three-strand hybrid allowed storing of planar drugs through their intercalation and, in addition, increased the lower critical temperature of the volume phase transition. In general, the control of the swelling–shrinking properties of this kind of hydrogels by setting an appropriate temperature program could be employed for the on–off pulsatile controlled drug release. The evaluation of properties, biocompatibility and cytotoxicity of novel nanogels on Hela and Insulinoma cells was done.
2. Materials and methods
2.1. Materials
N-Isopropyloacrylamide (NIPA 97%), N,N′-methylenebisacrylamide (BIS, 99.5%), potassium persulfate (KPS, 99.99%), acrylic acid (AAc 99%), fetal bovine serum (FBS), horse serum (HS), sodium hydroxide (NaOH), sodium chloride (NaCl), potassium chloride (KCl), monosodium phosphate (NaH2PO4), disodium phosphate (Na2HPO4), trypsin and dimethylforamide (DMF) were purchased from Sigma Aldrich (St. Louise, MO, USA). NIPA monomer was recrystallized twice from benzene and hexane mixture (1:9) to remove inhibit substances before polymerization of nanogels. Penicillin–streptomycin (10000 U mL−1), Gibco Fungizone®, RPMI 1640, DMEM, GlutaMAX™ media and MTT kits were provided by Thermo Fisher Scientific (Walthan, MA USA). Oligonucleotides (oligo1 and oligo2) modified by Acrydite® group were synthesized, standard-desalting purified and freeze-dried by Integrated DNA Technologies (IDT, Caralville, IA, USA). Two-part DNA linker (oligo3) was prepared by Friz Biochem (Neuried, Germany). The sequences and molecular masses of all oligonucleotides are given below:
• oligo1 5′-Acrydite-GGGGG-GCTCTTGGAACT-3′, MW: 5529.7, Tm = 57.3 °C.
• oligo2 5′-Acrydite GGGGG-TGAGTAGACACT-3′ MW: 5562.7, Tm = 53.0 °C.
• oligo3 5′-ACTCATCTGTGACGAGAACCTTGA-3′ MW: 7336.8, Tm = 56.0 °C.
Initial solutions of oligo1 and oligo2 (c = 238 μmol mL−1) were prepared in 0.01 M PBS buffer of pH = 7.4, while oligo3 (c = 330 μmol mL−1) solution was prepared in the same buffer with 0.15 M NaCl and 0.002 M KCl. Doxorubicin hydrochloride (Dox) was purchased from LC Laboratories (Woburn, MA, USA). Concentration of Dox in the stock solution (1 mL DMSO, 0.01 M PBS, pH 7.4) was determined by taking UV-Vis spectra; Dox extinction coefficient, ε485 nm = 10410 L mol−1 cm−1.33,62 For the preparation of all solutions, deionized water of conductivity 0.056 μS cm−1 (Milli-Q, Millipore) was used.
2.2. Synthesis of PNIPA-co-AAc nanoparticles (NPs) modified covalently with oligo1 and oligo2 strands
Neat- (without oligonucleotides) and PNIPA-co-AAc-oligo1-2 nanogels were synthesized using the surfactant-free emulsion polymerization method. NIPA, AAc and BIS monomers were dissolved in 9 mL of deionized water. The total concentration of NIPA, BIS and AAc equaled 76 mM mL−1. We have found that to get appropriate electrokinetic potential for optimal interaction of nanogels with the cell membranes and good effectiveness of the hybridization with oligo3 the amount of introduced AAc should be just 10% with respect to the concentration of NIPA monomer (68 mM). In the case of PNIPA-co-AAc-oligo1-2 nanogels, 50 μL of initial solutions of oligonucleotides (oligo1 and oligo2, respectively) were diluted separately in 3 mL of 0.01 M PBS (pH = 7.4) to achieve concentration 3.34 μmol mL−1. After 10 min stirring, both solutions were added to a 6 mL solution with monomers. The mixture was stirred at 1400 rpm for 1 h and heated under an argon atmosphere up to 70 °C, which value is higher than the melting point temperature, Tm, of each oligonucleotide. After 1 h, 10 mg 1 mL−1 KPS was added to initiate the free-radical polymerization. The reaction was continued for 4 h under argon atmosphere and at 1400 rpm stirring. Next, the solution was slowly cooled down to room temperature and kept at that temperature for 24 h. To remove the unbound residues, the nanogels were purified using the purified-water dialysis (Spectra/Por RC, MCWO 8–10 kDa) for 7 days. Water was exchanged 3 times per day. The effectiveness of the dialysis process was controlled by measuring conductivity of the dialysate solution until it reached a conductivity of pure, deionized water (0.056 μS cm−1). The yield of synthesis and the concentrations of bound oligonucleotides were estimated by measuring UV-Vis absorbance (at 270 nm) of nanogel solutions, as well as of dialysate solutions with residues. The value of extinction coefficient, ε270 nm, 166350 L mol−1 cm−1 was applied in the calculations. For the comparison purpose, we also synthetized PNIPA-co-AAc nanogels with separately introduced oligo1 and oligo2 strands.
2.3. Hybridization of oligo3 linker with strands in PNIPA-co-AAc-oligo1-2 NPs
3 mL of PNIPA-co-AAc-oligo1-2 NPs solution with oligo1-2 concentration 1.929 μmol mL−1 were heated slowly to 60 °C (a value close to Tm of each oligonucleotide) for 30 min to stretch the introduced strands and to start the shrinking process of PNIPA-co-AAc-oligo1-2 NPs. Simultaneously, the complementary (to both: oligo1 and oligo2) strand oligo3 was heated to the same temperature. The presence of 0.15 M NaCl and 0.002 M KCl in the solution helped in stabilization of the double stranded form of the three-strand hybrid and had no influence on the shrinking process of the polymer-based NPs. Subsequently, both solutions were mixed and incubated at 30 °C for 2 hours, where the swelling process of nanogels and the hybridization process took place. The concentration of oligo3 was by 30% higher than the sum of concentrations of oligo1 and oligo2 strands to maximize the hybridization yield. After 2 hours the solution was cooled down in an ice bath and then stored, for 24 h, in a fridge to complete the hybridization. After this step, the nanogels were additionally purified to remove all unbound species using the dialysis process with the control of conductivity of the dialyzed solution. The effectiveness of the dialysis process was controlled by measuring conductivity of the dialysate solution until it reached a conductivity of pure, deionized water (0.056 μS cm−1). The total concentration of all oligo1-2-3 species equaled 3.815 μmol mL−1 and was estimated spectrophotometrically (at 260 nm). The corresponding extinction coefficient, ε260 nm, was determined to be 200225 L mol−1 cm−1 and was used for nanogels dissolved in 0.01 M PBS buffer with 0.15 M NaCl and 0.002 M KCl. For the comparison purpose, we also added oligo3 linker to alone PNIPA-co-AAc-oligo1 and PNIPA-co-AAc-oligo2 nanoparticles.
2.4. Doxorubicin loading
The drug-loaded nanogels were prepared by mixing 2 mL of 104.4 μg mL−1 Dox solution with 2 mL of suspensions of particular nanogels (7.8 mg mL−1, with coligo1-2 = 1.929 μmol mL−1 or coligo1-2-3 = 3.815 μmol mL−1). The concentration of Dox just after mixing with nanogels equaled 52.2 μg mL−1. Concentrations of Dox in particular nanogels were determined spectrophotometrically (ε485 nm = 10410 L mol−1 cm−1,33,62). The mixture was kept, for 24 h, at room temperature in the dark and was gently stirred to allow Dox to be absorbed. Nanogels with bound Dox were separated by employing centrifugation (60000 rpm for 90 min). The completeness of purification was controlled spectrophotometrically (λ = 485 nm). Finally, the NP sediments were suspended in 4 mL of 0.01 M PBS of pH 7.4.
2.5. In vitro doxorubicin release
2 mL of each Dox-loaded nanogel solution with a known drug concentration were placed in a dialysis bag (MWCO = 10 kDa). The bags were dialyzed against 25 mL of 0.01 M PBS with 0.15 M NaCl and 0.002 M KCl (pH = 7.4 or 5.5). We used the release medium with appropriate amount of ions to keep constant ionic strength of the working solution; the point was not to promote the additional matrix hydration and the erosion of the hydrogel-based PNIPA-co-AAc-oligo1-2 NPs. Gentle stirring (200 rpm) was applied during the process. The released Dox was determined in the solution outside the dialysis bag in a flow-cell system (sampling time = 1 min). We have performed the measurements either at constant temperatures: 37 °C and 45 °C (they are routinely used in the hyperthermia therapy), or the temperature was oscillating between 37 and 45 °C. The percent drug release was calculated using the following equation:where At is absorbance of the drug present in the solution at particular sampling time and At0 is absorbance of drug initially present in the vesicles.

2.6. Characterization of nanogels
2.7. Cell culture
Human Adenocarcinoma cervicis uteri Hela cells were cultured at 37 °C in RPMI 1640 medium with GlutaMAX™ supplement and a mixture of 10% FBS, 100 μg mL−1 streptomycin, and 100 U mL−1 penicillin in humidified atmosphere containing 5% of CO2. Human Insulinoma β-TC3 cell line was maintained at 37 °C in Dulbecco's modified Eagle's medium (DMEM), high glucose GlutaMAX™, 12% HS, 3% FBS, 10 μg mL−1 fungizone, 100 μg mL−1 streptomycin and 100 U mL−1 penicillin in humidified atmosphere containing 5% CO2.2.8. In vitro cell viability assays
The cytotoxicity of Dox/PNIPA-co-AAc-oligo nanogels against Hela and β-TC3 cancer cell lines was assessed by MTT plate-based assay. Briefly, the cells were seeded onto 96-well plates. The density of β-TC3 cells was 3000 cells/100 μL per well and of Hela cells was 5000 cells/100 μL per well. Cells were incubated at 37 °C in 5% CO2 atmosphere for 24 h to allow the cell attachment. On the day of experiments, the cells were washed with a warm PBS. Next, cells were incubated with free Dox, Dox/PNIPA-co-AAc, Dox/PNIPA-co-AAc-oligo1-2 and Dox/PNIPA-co-AAc-oligo1-2-3. Dox concentrations ranged from 0.05 to 20 mg mL−1, and the nanogel concentrations was constant: 7.8 mg mL−1. As the control, particular unloaded nanogels were incubated. The cells were incubated for 30 min or 2 h intervals at 37 °C, washed three times with PBS (100 μL) and fresh growth media was added. The cells were incubated for a total of 96 h. For MTT tetrazolium assay, 25 μL of 5 g mL−1 solution of MTT reagent was added and incubated for 2 h. Next, 100 μL of lysis buffer (20% SDS, 50% DMF, pH 4.5) were introduced. After addition of the probes and targets of particular assays, the fluorescence (fluorescence excitation and emission at 480 and 520 nm) and absorbance (at 570 nm, reference wavelength 630 nm) were measured with a POLARStar Omega plate reader (BMG Labtech, Aylesbury, UK). All experiments were repeated four times. All fluorescence results presented in this work were corrected vs. the values obtained for media without cells and with and without dyes and the values estimated after interaction of assays constituents with Dox unloaded particular oligonucleotide-based nanogels.2.9. Cellular-uptake estimation by flow cytometry
Flow cytometry (FCM) was used to determine Dox uptake capability of the PNIPA-co-AAc-oligo nanoparticles into particular cell lines. After appropriate time of incubation, Hela or β-TC3 cells with free Dox, Dox/PNIPA-co-AAc, Dox/PNIPA-co-AAc-oligo1-2, Dox/PNIPA-co-AAc-oligo1-2-3 with Dox and blank nanogels without Dox were transferred from well-plates into falcon-type centrifuge vials. The cells were washed twice with PBS buffer to remove residual nanoparticles. After treatment with 0.25% trypsin solution the adhered cells were harvested, washed three times with PBS buffer, the media were removed and finally all cells were suspended in 1 mL of PBS in falcon vials. The Dox fluorescence was recorded with a flow cytometer (Attune®, Applied Biosystems, Life Technologies, Carlsbad, CA) after selected times.2.10. Confocal microscopy assay
The fact of intracellular localization of free Dox and Dox from particular nanogels was visualized with a confocal laser scanning microscope (CLSM, Olympus FV1000, Olympus, Hamburg, Germany). The cells were seeded onto 6-well plates and cultured with concentrations of: (a) 10 mg mL−1 free Dox and (b) the same Dox concentration, where Dox was immobilized in particular NPs, with coligo1-2 = 1.929 μmol mL−1 and coligo1-2-3 = 3.815 μmol mL−1, respectively. After particular time of an incubation the cells were washed with PBS and fixed with 5% paraformaldehyde solution in PBS. One drop of Canadian Balsam was placed on each slide to seal the cell samples. Finally, the images of the stained probes were taken. Doxorubicin was excited with 488 nm wave and the emission was recorded at 560–600 nm.3. Results and discussion
3.1. Synthesis and physicochemical characterization of NPs
Fig. 1 presents the stepwise preparation of novel nanoparticles: PNIPA-co-AAc-nanogels with three-strand hybridized oligonucleotides. In the first step (see Fig. 1A), PNIPA-co-AAc-oligo1-2 nanogels were synthesized by free-radical and surfactant-free emulsion copolymerization of NIPA, AAc and BIS monomers37 and oligo1 and oligo2 strands were bound covalently to the gel net. The second step (Fig. 1B) involved the hybridization of the oligo3 linker that was 50% complementary to both: oligo1 and oligo2 strands. Finally, the oligo1-2-3 three-strand hybrids were formed. The obtained delivery system, PNIPA-co-AAc-oligo1-2-3 NPs, was compared with PNIPA-co-AAc- and PNIPA-co-AAc-oligo1-2 NPs. We did appropriate simulation to make sure the probability of hybridization between 1 and 2 oligonucleotides was low, see Fig. 1S–3S in (ESI†). Moreover, the probability of stronger interactions, that is of hydrogen bonding and Hoogsten interactions, between combinations of particular nucleic bases during the polymerization process of PNIPA-co-AAc-oligo1-2 NPs was significantly lower at 70 °C then at physiological temperature of 37 °C. In the case of hybridization of oligo3, oligo2 and oligo1 in PNIPA-co-AAc-oligo1-2 NPs we noticed that cooling of the medium to 4 °C allowed us to increase the efficiency of hybridization of the oligo-1-2- and oligo-3 species in PNIPA-co-AAc-based nets. A comparison of mean particle sizes (hydrodynamic diameters, Dh) and zeta potentials (ζ) of all employed types of the nanogels is presented in Table 1 and Fig. 4S and 5S in ESI;† both parameters were determined in pure H2O at 25 °C, 37 °C and after shrinking process at 45 °C by using the dynamic light scattering method (DLS).38,39 It is seen, that the introduction of oligo1 and oligo2 strands into PNIPA-co-AAc NPs and the hybridization of the strands have a significant influence on the size and zeta potential at 37 and 45 °C. We would like to stress that before the shrinking process, both oligonucleotide-based nanogels existed as well-suspended and optically transparent colloidal solutions. For comparison, the PNIPA-co-AAc NPs solutions were more turbid. Interestingly, the lyophilized forms of all examined nanogels regained their initial properties after dispersion in distilled water. | ||
| Fig. 1 (A) Scheme of first step of synthesis of PNIPA-co-AAc-oligo1-2-3 nanogels: a covalent introduction of oligo1 and oligo2 strands during polymerization of PNIPA-co-AAc-based NPs. (B) Second step of synthesis of PNIPA-co-AAc-oligo1-2-3 nanogels; hybridization process of linker oligo3 with oligo1-2 in PNIPA-co-AAc-oligo1-2 NPs. | ||
| Nanogel type | PNIPA-co-AAc | PNIPA-co-AAc-oligo1-2 | PNIPA-co-AAc-oligo1-2-3 |
|---|---|---|---|
| Size at 25 °C (nm) | 495.8 ± 11.1 | 481.7 ± 13.5 | 440.1 ± 12.3 |
| Size at 37 °C (nm) | 255.5 ± 8.5 | 246.6 ± 7.5 | 212.6 ± 6.0 |
| Size at 45 °C (nm) | 214.6 ± 4.5 | 165.4 ± 5.0 | 93.0 ± 3.5 |
| Zeta at 25 °C (mV) | −11.3 ± 0.3 | −9.9 ± 0.5 | −15.6 ± 0.7 |
| Zeta at 37 °C (mV) | −22.4 ± 0.7 | −24.1 ± 0.9 | −25.9 ± 0.8 |
| Zeta at 45 °C (mV) | −30.1 ± 0.5 | −25.5 ± 0.4 | −28.8 ± 0.7 |
| PdI 25 °C/37 °C/45 °C | 0.21/0.11/0.10 | 0.25/0.18/0.16 | 0.28/0.15/0.13 |
Fig. 2 depicts regular photos and SEM and TEM micrographs of all examined types of nanogels. Apparently, solutions of plain nanogel and nanogel with incorporated oligonucleotides differed much in clarity at room temperature, see Fig. 2A. After introduction of oligo3 linker, through hybridization with oligo1 and oligo2 in PNIPA-co-AAc-oligo1-2 nanogels, the optical clarity remained very good. We tie a better clarity of solutions of NPs with oligonucleotide strands with the absorption of more water molecules by the gel nanoparticles.40
 | ||
| Fig. 2 (A) Comparison of appearance of PNIPA-co-AAc nanogel solution (left) and PNIPA-co-AAc nanogels with immobilized oligo1 and oligo2 strands (right), T = 25 °C. (B) SEM micrographs of typical PNIPA-co-AAc-oligo1-2 nanogels (left) and PNIPA-AAc-oligo1-2-3 NPs (right), both obtained after gentle lyophilisation of NPs solution. (C) TEM micrographs of PNIPA-co-AAc NPs. (D) TEM micrographs of PNIPA-co-AAc-oligo1-2 and (E) TEM pictures of PNIPA-co-AAc-oligo1-2-3 nanogels. | ||
The difference in morphology of lyophilized PNIPA-co-AAc-oligo1-2 nanogels and the corresponding hybrids is presented in Fig. 2B. The shapes of unmodified PNIPA-co-AAc- (not shown) and PNIPA-co-AAc-oligo1-2 nanogels are almost identical and are more spherical and homogenous in comparison to PNIPA-co-AAc-oligo1-2-3 NPs. The sizes of nanogels presented in Fig. 2B are also very similar. Since all SEM samples were just gently lyophilized, a part of the crystallized water was not removed from nanogels. Thus, the sizes of the nanogels presented in the figure correspond rather to their partially-swollen state. More details on the morphology of all nanogels could be gained from the TEM pictures, see Fig. 2C–E. Before taking TEM pictures the gel samples were dried at 50 °C. Thus, the amount of water in the nanogels should be much lower than at lyophilized samples and finally the TEM pictures better reflect the nanogel size of the shrunken state. Generally, the PNIPA-co-AAc-oligo1-2-3 NPs always had the smallest sizes. Interestingly, we also noticed, that plain PNIPA-co-AAc NPs possessed smoother surface than the nanogels modified with oligonucleotides. To visualize the surface of nanogels, we used uranyl acetate (UA) which strongly electrostatically interacts with negatively charged phosphate groups of oligonucleotides and has stronger affinity to negatively charged phosphate groups compared to carboxylic groups that are also present in nanogel nets.41,42 As it can be seen in Fig. 2D, the surface of PNIPA-co-AAc-oligo1-2 NPs is better defined, darker (higher amount of UA was adsorbed uniformly in the net) and uneven. After a 28 h procedure of incubation of nanogel samples in an UA solution the UO22+ cations filled completely the nanoparticles, which was reflected by black color of the nanogels. The presence of single DNA strands guaranteed the uniform distribution of UO22+ in the nanoparticles (Fig. 2D). For plain nanoparticles the concentration of UO22+ was apparently lower at the surface (this was a result of rinsing with water). An interesting situation appeared in the case of PNIPA-co-AAc-oligo1-2-3 NPs. Double stranded DNA was formed at the surface and this apparently resulted in a lower content of the UO22+ cations in the outer layer of the nanoparticles after rinsing (see Fig. 2E). Contrary, at the center of PNIPA-co-AAc-oligo1-2-3 NPs, the black color was intensive, so it can be concluded that the hybridization process was rather limited there.
Next, a comparison of the SEM and TEM pictures with the mean particle sizes obtained from the DLS measurements was done. The mean sizes determined in pure H2O by dynamic light scattering method (DLS) for nanogels solutions in swelled (25 °C)-, partially shrunken (37 °C)- and shrunken form of the nanogel particles (45 °C) are presented in Table 1. After comparing the SEM, TEM and DLS results (various conditions of these technique's were taken into account) it can be said that the sizes measured by SEM better agreed with the DLS sizes obtained at 37 °C. Contrary, the TEM results better reflected the DLS sizes obtained for the shrunken form at 45 °C. These findings make sense, since the samples before the SEM examination were partially/gently lyophilized and the size of the swollen particles in the solution was preserved. Contrary, those particles that were examined with TEM were completely dried before the measurements and finally their size was similar to the size of shrunken particles in the solution.
The changes in mean sizes of all types of examined nanogels correlated somewhat with the changes in zeta potential, see Table 1. A significant decrease in the negative value of zeta potential was noticed for PNIPA-co-AAc-oligo1-2-3 NPs after the three-strand hybridization. Since most of biological cells have negative zeta potentials, nanobiomolecules should also be of slightly negative zeta potentials. As a result, they do not stick electrostatically to cell membranes; they interact through a receptor. The receptor–ligand bond should be strong enough to overcome a modest electrical repulsion.43
Typical FT-IR spectra of gently lyophilized PNIPA-co-AAc-, PNIPA-co-AAc-oligo1-2- and PNIPA-co-AA-oligo1-2-3 NPs are presented in Fig. 3.
 | ||
| Fig. 3 FTIR spectra of PNIPA-co-AAc, PNIPA-co-AAc-oligo1-2 and PNIPA-co-AAc-oligo1-2-3 nanoparticles. | ||
In all spectra, the bands characteristic for NIPA monomer can be seen.44 The infrared peak at circa 3450 cm−1 corresponds just to PNIPA and PNIPA-co-AAc lattices. It is a result of interactions of the gel net with water molecules. The intensity of this peak indicates the amount of water in the polymerized gels that remained in the nets after the lyophilization process. The presence of oligonucleotides, and especially of hybridized strands, led to a significant reduction of intensity of this peak. Apparently, the hybridization led to partial removal of water from the nanoparticles.45,46 The strong peaks placed at 1649.5 and 1433.5 cm−1 correspond to the ![[double bond splayed left]](https://www.rsc.org/images/entities/char_e009.gif) C
C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and CO carbonyl stretching vibrations in NIPA.44 The presence of acrylic groups in the nanogel nets is indicated by the peak placed at 1718 cm−1 (the CO stretching vibration). The peak placed at 918 cm−1 that appears in the spectra of PNIPA-co-AAc-oligo1-2 and PNIPA-co-AAc-oligo1-2-3 corresponds to PO32− symmetric stretching vibration in introduced nucleotides.
C and CO carbonyl stretching vibrations in NIPA.44 The presence of acrylic groups in the nanogel nets is indicated by the peak placed at 1718 cm−1 (the CO stretching vibration). The peak placed at 918 cm−1 that appears in the spectra of PNIPA-co-AAc-oligo1-2 and PNIPA-co-AAc-oligo1-2-3 corresponds to PO32− symmetric stretching vibration in introduced nucleotides.
The presence of oligonucleotides bound to the gel network was also confirmed by UV-VIS spectra. The measurements were done after the dialysis. The corresponding DNA band appeared at circa 260 nm, see Fig. 4. The formation of three-strand strands can be concluded from the plots shown in Fig. 4A. The introduction of the third strand, complementary to the first and the second strands, resulted in a double increase in DNA band intensity. This clearly indicated the three-strand hybridization, since when the gel contained just one strand (e.g. oligo1) the hybridization with linker oligo3 resulted in absorbance increase by less than 50%. Fig. 4B presents changes in absorbance recorded during the shrinking process of the hybridized PNIPA-co-AAc-oligo1-3 and PNIPA-co-AAc-oligo1-2-3 nanogels. It can be seen that absorbance of oligo1-2-3 hybrid (λ = 260 nm) increased less (smaller turbidity) compared to PNIPA-co-AAc-oligo1-3. This may be explained by more complete dehydration of PNIPA-co-AAc-oligo1-3 nanogels.
 | ||
| Fig. 4 (A) UV-Vis spectra of PNIPA-co-AAc-oligo1-2- (red, a), PNIPA-co-AAc-oligo1-3- (green, b) and PNIPA-AAc-oligo1-2-3 nanogels (blue, c). (B) UV-VIS spectra for PNIPA-co-AAc-oligo1-2-3 NPs (blue) and PNIPA-co-AAc-oligo1-3 NPs (green) at various temperatures. | ||
The electrochemical impedance spectroscopy (EIS) is widely applied for the detection of dsDNA and analysis of the hybridization process of single stranded oligonucleotides. An estimation of the impedance, as well as charge transfer resistance, capacity and conductivity parameters can be determined for layers of oligonucleotide strands.47,48 The diameter of the semicircle in the corresponding Nyquist plots is a measure of the electron transfer resistance at the electrode surface, RCT (see inset in Fig. 5).
 | ||
| Fig. 5 EIS Nyquist plots obtained for 2 mM [Fe(CN)6]3−/4−] in 0.01 PBS, pH 7.4 solution for deposited NP layers on GC electrode. (a) PNIPA-co-AAc-1-2- (red), (b) PNIPA-co-AAc-1-3- (green) and (c) NIPA-co-AAc-1-2-3 (blue) nanogels, φ = 3 mm, T = 37 °C. Inset: Randles circuit used for fitting simulated plots (solid lines in figure) into experimental data. Rs – solution resistance, RCT – charge transfer resistance, CPE – constant phase element and W – Warburg impedance. | ||
For the nanogel layers that are in good contact with the electrode surface, a decrease in double layer capacitance should take place and the barrier for interfacial electron transfer should increase. The EIS spectra were obtained to confirm the presence of the helix – the product of DNA three-strand hybridization in nanogels. The EIS results for PNIPA-co-AAc-oligo1-2-, PNIPA-co-AAc-oligo1-3- and PNIPA-co-AA-oligo1-2-3 NPs, at 20 °C are presented in Fig. 5. Since the length of the obtained helix was different in the considered cases, different EIS spectra should be obtained. All obtained Nyquist plots contained a semicircle and a linear component. The presented results were acquired at the formal potential of equimolar mixture of Fe(CN)6]3−/4− redox couple. As it can be seen in Fig. 5, the diameter of EIS semicircles (equal to RCT) grew in order: PNIPA-co-AAc-oligo1-2-, PNIPA-co-AAc-oligo1-3- and PNIPA-co-AA-oligo1-2-3 NPs. Apparently, RCT increased with length of hybridized part of oligonucleotides and was the highest for the longest dsDNA (-oligo1-2-3). It can be interpreted as the result of an increase in electrostatic repulsions between the redox probe and the increased amount of the negatively charged phosphate groups in longer hybrids. To calculate the EIS parameters we have employed the Ershler–Randles equivalent circuit (see inset in Fig. 5) in the fitting process. In this circuit the double layer capacitance is replaced by constant phase element (CPE). The lines in Nyquist plots fit well the experimental data what confirms the correct selection of the equivalent circuit.36,49,50 The determined impedance parameters and contact angle values are listed in Table 2. The formulas used in the determination of all EIS parameters are presented in the ESI.† As it was expected, the solution resistance, Rs, had similar values for all measurements and was independent on the hybridization length of the oligonucleotides. What is important, the mass transport to the electrode of redox probe is not hindered in all nanogels layers, so the Warburg parameter is nearly constant. The double layer capacitance decreases with increasing length of the duplex. The exponent parameter ϕ is smaller than 1, while for monocrystalline or liquid electrodes of purely capacitive behavior it is equal to 1. Thus, constant phase element indicates a rather not ideal capacitor behavior.49
| PNIPA-co-AAc-oligo1-2 | PNIPA-co-AAc-oligo1-3 | PNIPA-co-AAc-oligo1-2-3 | |
|---|---|---|---|
| Rs (Ω) | 309 ± 5.2 | 312 ± 4.1 | 305 ± 3.7 |
| RCT (Ω) | 2614 ± 12.4 | 5843 ± 10.7 | 7543 ± 13.1 |
| Cdl (μF) | 0.799 ± 0.05 | 0.497 ± 0.03 | 0.393 ± 0.03 |
| Φ | 0.799 ± 0.06 | 0.822 ± 0.07 | 0.834 ± 0.05 |
| σ (Ω rad1/2 s−1/2 cm2) | 80.4 ± 0.7 | 78.2 ± 0.4 | 83.1 ± 0.5 |
| Contact angle (°) | 49.60 | 52.12 | 68.84 |
Next, we made sure that all nanogels are thermoresponsive, see Fig. 6. Fig. 6A presents normalized changes of hydrodynamic diameters (Dh) plotted vs. temperature. The sudden decrease of diameters is related to the shrinking process that takes place in all examined nanogels.37 In DLS technique, the information about the changes in the dynamics of nanoparticles is derived from the second order autocorrelation function, the decay of which is related to size (Dh) and therefore to diffusion coefficient (D) of the nanoparticles. These parameters are linked in the Stokes–Einstein eqn (1):
 | (1) |
 | ||
| Fig. 6 (A) Plots of hydrodynamic diameter, Dh, (normalized vs. hydrodynamic diameter at 25 °C) vs. temperature obtained for PNIPA-co-AAc- (black circle), PNIPA-co-AAc-oligo1-3- (green, star), PNIPA-co-AAc-oligo1-2- (red, triangle) and PNIPA-co-AAc-oligo1-2-3 (blue, square) nanoparticles. Inset: hydrodynamic diameter plotted vs. total concentration of introduced oligonucleotides at 37 °C. (B) Changes of zeta potential, ζ, vs. temperature obtained for: PNIPA-co-AAc-, PNIPA-co-AAc-oligo1-2- and PNIPA-co-AAc-oligo1-2-3 NPs. Inset: plot of zeta potential vs. total concentration of introduced oligonucleotides obtained at 37 °C. Oscillating changes of hydrodynamic diameters (C) and zeta potential (D) measured in three-point temperature system (37 °C to 45 °C and back to 37 °C) and visualized for three consecutive cycles. Interval time 5 min. | ||
For nanogels with oligo1-2-3 hybrids the decrease in size started at temperatures slightly higher than for other nanogels. At 37 °C, the extent of the shrinking process for PNIPA-co-AAc-, PNIPA-co-AAc-oligo1-2-, PNIPA-co-AAc-oligo1-3- and PNIPA-co-AA-oligo1-2-3 NPs was 87, 75, 65 and 45%, respectively.
The differences in size measured at 37 °C vs. oligonucleotide concentration (that is related to total concentration of oligo1-2- and oligo1-2-3 strands introduced into nanogel nets) are presented in the inset of Fig. 6A. After the hybridization of oligo3 with oligo1 and oligo2 strands in PNIPA-co-AAc nanogels, we observed a significant decrease in nanogel size. This, once again confirmed that we observed the formation of oligo1-2-3 hybrids in the nanogel nets. We also plotted zeta potential changes vs. temperature, see Fig. 6B. We noticed a decrease in zeta potential that was correlated with the decrease in size of the nanogels during their shrinking processes. An interesting linear increase of zeta potential was noticed at 37 °C in the dependence on oligonucleotide concentration, see inset in Fig. 6B. This reversible thermal switching of size and zeta potential was observed for all nanoparticles upon cycling temperature between 37 and 45 °C. It is important to stress, that the oscillation of temperature can generate the shrinking process and the conformational changes in duplex DNA, but not the denaturation process.51,52 These predenaturation changes can promote the release of an intercalator drug from the DNA/hydrogel based lattices.19 Its worth of noting that the characters of Dh and zeta potential (ζ) changes were contradictory. Zeta potential was depressed with temperature while the values of Dh were enhanced by the hybridization.
Independently, we looked closer at the absorbance changes at 260 nm after a sudden increase of temperature. The changes in absorbance vs. time (temperature decreased) are presented in Fig. 7A and B. These changes reflected the occurrence of two processes: (a) conformation change/denaturation of the three-strand hybrid and (b) shrinking of the nanogels. In the case of Fig. 7A, 0.5 mL of hot supporting solution (0.1 M PBS, pH 7.4, T = 75 °C) was added to 0.5 mL of a gel solution. The final temperature was 45 °C. We noticed a very fast and discontinuous shrinking of all examined nanogels after addition of the hot solution. Then, in time, the swelling of all nanogels could be seen. In the case of PNIPA-co-AAc NPs the initial absorbance was relatively high, as the nanogel solutions were in general more turbid. We noticed that the swelling process of all nanogels was reversible and was completed in 10 min for PNIPA-co-AAc- and PNIPA-co-AAc-oligo1-2 NPs and in 8 min for PNIPA-co-AAc-oligo1-2-3 NPs. A 50% decrease in absorbance recorded after completing of the process was a result of dilution of gel samples. The presence of oligo1-2-3 hybrid resulted in faster phase transformations.
 | ||
| Fig. 7 (A) A vs. t obtained from UV-Vis experiments. PNIPA-co-AAc- (black, solid line), PNIPA-co-AAc-oligo1-2- (red line) and PNIPA-co-AAc-oligo1-2-3 NPs (blue line) after adding of 0.5 mL of hot water (75 °C) to 0.5 mL of solution with particular nanogel (25 °C). Temperature after mixing of solutions equaled 45 °C. (B) A vs. t obtained from UV-Vis experiments. Oligo1-2-3 hybrid dissolved in 0.1 M PBS of pH 7.4 (pink line) and PNIPA-co-AAc-oligo1-2-3 NPs (blue line). 1 mL of particular solution was heated for 10 min at 70 °C. Rate of heating: 2.3 °C per 1 min, accuracy ± 0.2 °C. | ||
It cannot be clearly concluded from Fig. 7A that the gel net protects dsDNA vs. possible denaturation process at applied higher temperature, as the temperature of 45 °C is the temperature where typical denaturation process is not well seen in UV-Vis measurements. Thus, only conformational change at this temperature should be possible after shrinking and then reversible swelling of DNA-hydrogel-based nanogel composites.52 Fig. 7B presents a plot of A vs. t obtained with UV-Vis spectroscopy for the oligo1-2-3 hybrid and PNIPA-co-AAc-oligo1-2-3 NPs after heating of undiluted particular solution to 70 °C. The results confirmed the idea that the gel net can enable the regeneration of the double stranded form of oligo1-2-3 hybrid, while in the solution environment, the denaturation process was irreversible. Moreover, consecutive heating and cooling cycles of PNIPA-co-AAc-oligo1-2-3 NPs gave similar results as those presented in Fig. 7B. When the heating was limited to 45 °C, only a change in DNA conformation and the gel shrinking process could take place. This apparently led to moderate release of selected accumulated drug-intercalator, doxorubicin.
3.2. Loading efficiency and temperature dependent release kinetics
The evaluation of the potential of PNIPA-co-AAc-, PNIPA-co-AAc-oligo1-2-, and PNIPA-co-AAc-oligo1-2-3 nanogels as drug carriers for cancer therapy was investigated by performing in vitro release measurements, see Fig. 8. For the investigation a model intercalating drug, anthracycline doxorubicin (Dox), was selected. This drug was used in several-types of delivery nanosystems in clinical cancer therapy.53,54 It is known, that the interaction of planar Dox molecule with dsDNA can involve two types of noncovalent interactions: the strong one (intercalation) and the weak one (electrostatic).55 The existence of the environment in a gel form can influence the values of the binding constants, promotion of the intercalation and the releasing process.51,52 The loading efficiency of Dox into PNIPA-co-AAc-, PNIPA-co-AAc-oligo1-2-, and PNIPA-co-AA-oligo1-2-3 nanogels was determined from UV-Vis measurements data and is presented in Fig. 8A. The release experiments were done in pH 6.5 corresponding to intratumor interstitial conditions. The accumulation was done in a 51.91 μg mL−1 solution of Dox. The highest accumulation of Dox drug took place in PNIPA-co-AAc-oligo1-2-3 NPs. The percent of encapsulation of doxorubicin equaled 30.2, 40.3 and 67.6%, for PNIPA-co-AAc-, PNIPA-co-AAc-oligo1-2-, and PNIPA-co-AAc-oligo1-2-3 NPs, respectively. We used the McGhee and von Hippel model for the determination of the parameters of binding between oligonucleotides and Dox.56,57 The formula describing the interactions between DNA chains and the planar compound Dox is given as eqn (2),
 | (2) |
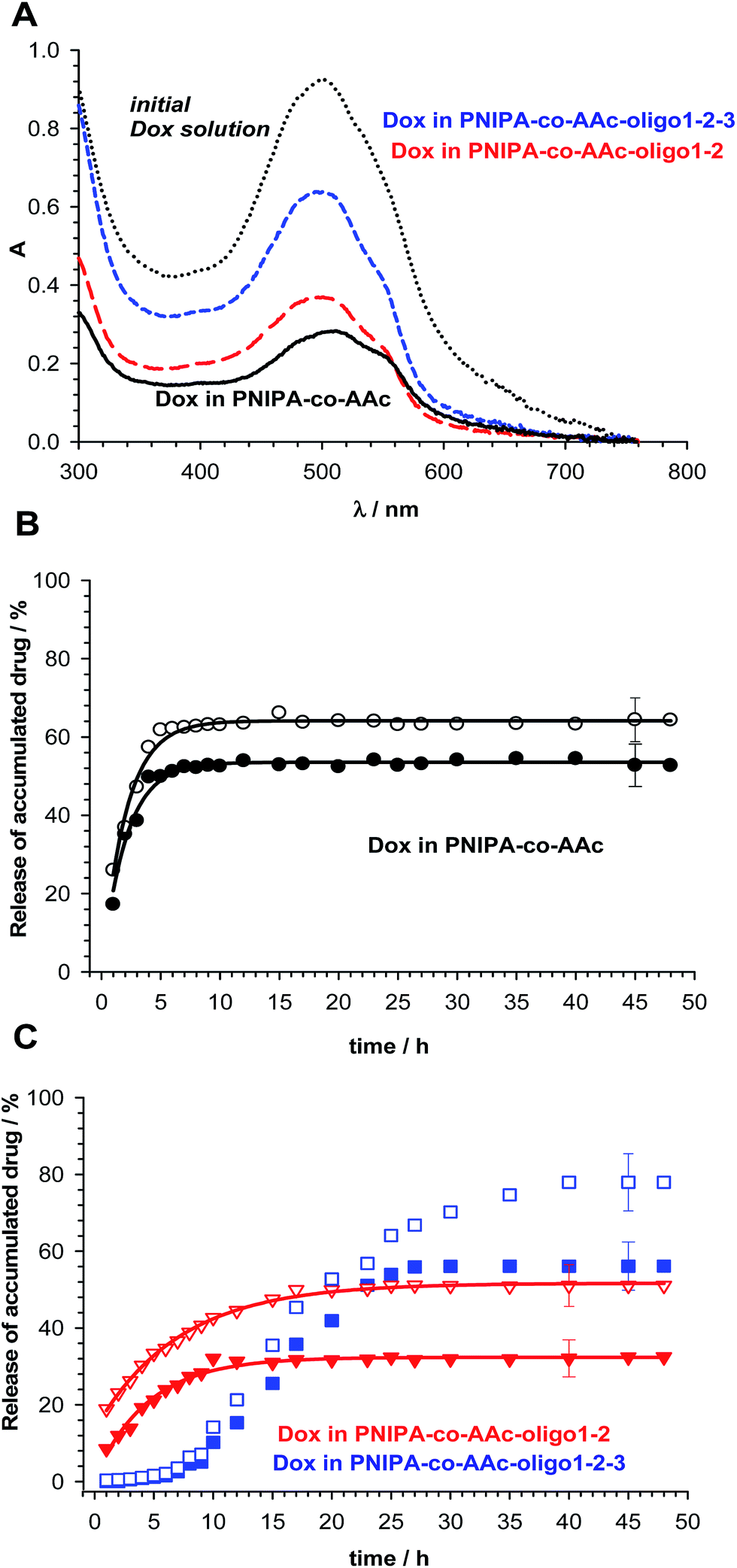 | ||
| Fig. 8 (A) UV-Vis spectra recorded for initial concentration of Dox used for accumulation in nanogels (black, dotted line) and Dox encapsulated in PNIPA-co-AAc- (black, solid line), PNIPA-co-AAc-oligo1-2- (red line) and PNIPA-co-AAc-oligo1-2-3 NPs (blue line). (B) Dox release profiles obtained from UV-Vis measurement data for PNIPA-co-AAc nanogels at 37 (filled circles) and 45 °C (empty circles). (C) Dox release profiles obtained from UV-Vis measurement data for PNIPA-co-AAc-oligo1-2- (triangles, red) and PNIPA-co-AAc-oligo1-2-3 NPs (squares, blue) at 37 (filled points) and 45 °C (empty points). | ||
Doxorubicin was released from the investigated nanogels at 37 and 45 °C. The influence of temperature oscillating between 37 and 45 °C was examined. The results are presented in Fig. 8B and C. At 45 °C the % of released Dox increased to circa 60, 50 and 80% of accumulated initial Dox amount for PNIPA-co-AAc-, PNIPA-co-AAc-oligo1-2, and PNIPA-co-AAc-oligo1-2-3 NPs, respectively (Fig. 8B and C). Surprisingly, we noticed a change in the way Dox was released from PNIPA-co-AAc-oligo1-2-3 nanogels compared to two other nanoparticles. The release process of Dox was slower; however, the final efficiency of Dox release was higher.
The process of Dox release from particular investigated nanogels was analysed with application of the empirical power Korsmeyer–Peppas model. The model is described by eqn (3):
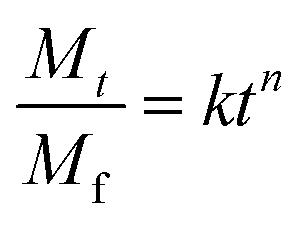 | (3) |
| Nanogel type | Release parameters of Dox | ||
|---|---|---|---|
| k | n | r | |
| 0–5 hours (37 °C/45 °C) | |||
| PNIPA-co-AAc | 0.46 ± 0.01/0.38 ± 0.01 | 0.46 ± 0.02/0.35 ± 0.02 | 0.998/0.998 |
| PNIPA-co-AAc-oligo1-2 | 0.51 ± 0.02/0.26 ± 0.01 | 0.70 ± 0.04/0.35 ± 0.02 | 0.995/0.993 |
| PNIPA-co-AAc-oligo1-2-3 | 0.97 ± 0.03/0.65 ± 0.02 | 1.35 ± 0.07/0.90 ± 0.05 | 0.996/0.996 |
|
|||
| 6–12 hours (37 °C/45 °C) | |||
| PNIPA-co-AAc | — | — | — |
| PNIPA-co-AAc-oligo1-2 | 0.46 ± 0.07/0.39 ± 0.08 | 0.45 ± 0.07/0.38 ± 0.09 | 0.975/0.979 |
| PNIPA-co-AAc-oligo1-2-3 | 3.06 ± 0.10/3.05 ± 0.20 | 3.29 ± 0.19/3.28 ± 0.20 | 0.997/0.993 |
|
|||
| 13–25 hours (37 °C/45 °C) | |||
| PNIPA-co-AAc | — | — | — |
| PNIPA-co-AAc-oligo1-2 | — | — | — |
| PNIPA-co-AAc-oligo1-2-3 | 1.90 ± 0.20/2.21 ± 0.10 | 1.04 ± 0.17/1.60 ± 0.08 | 0.988/0.996 |
|
|||
| 26–48 hours (37 °C/45 °C) | |||
| PNIPA-co-AAc | — | — | — |
| PNIPA-co-AAc-oligo1-2 | — | — | — |
| PNIPA-co-AAc-oligo1-2-3 | —/0.42 ± 0.03 | —/0.25 ± 0.02 | —/0.987 |
3.3. Cell viability assays
The evaluation of biocompatibility of nanoparticles was the first step in evaluation of their usefulness in drug delivery nanosystems. The cytotoxicity of investigated nanogels, PNIPA-co-AAc-, PNIPA-co-AAc-oligo1-2-, and PNIPA-co-AAc-oligo1-2-3 NPs, was assessed by doing MTT assay after an incubation of Hela and Insulinoma cell lines for 48 h. The concentration of nanoparticles equaled 7.8 mg mL−1. As it is shown in the inset of Fig. 9 all investigated nanogels possessed excellent biocompatibility and low cytotoxicity even at high concentrations. As the polymerization reaction of nanogels occurred without the application of surfactant agent and all PNIPA-based nanoparticles were of high purity, there existed a minor effect of growth of cells for plain PNIPA-co-AAc NPs.60 The percent of viability of Hela and Insulinoma cells determined after 48 h of their incubation at 37 °C with particular concentrations of unloaded Dox and Dox incorporated in PNIPA-co-AAc-, PNIPA-co-AAc-oligo1-2-, and PNIPA-co-AAc-oligo1-2-3 NPs are presented in Fig. 9A and B. We noticed that the lowest proliferation of cells took place for PNIPA-co-AAc-oligo1-2-3 nanogels with three-strand oligonucleotide hybrids. This meant that under given conditions the amount of Dox that penetrated particular cancer cells and therefore intercalated cell DNA was the highest. The final effect was slightly higher in the case of Hela cells. As at 37 °C, 50% of Dox remained bound in the oligo-1-2-3 nanogel matrix (see Fig. 9) we compared the cell viability results with the measurements done at 45 °C for the highest concentration of Dox (15 μg mL−1) where the effectiveness of the drug treatment was the highest. A comparison of the results obtained at 37 and 45 °C indicated that at 45 °C a decrease in cells viability took place. The magnitude of the decrease was circa 14 and 10% for PNIPA-co-AAc-, 24 and 19% for PNIPA-co-AAc-oligo1-2- and 40 and 36% for PNIPA-co-AAc-oligo1-2-3 NPs for Hela and Insulinoma cells, respectively. It should be stressed here, that generally in MTT test the viability of cancer cells is somewhat biased due to the final destination of the MTT dye and its metabolism done by dehydrogenase enzymes in cells' mitochondria. For most viable cells the mitochondrial activity is constant, so a change in total mitochondrial activity allows the estimation of the number of viable cells.61 The above findings might be influenced by the fact that the cell lines were investigated in two differed culture media. It is known, that the selected by us media provide high effectiveness of cell proliferation; however, the differences in their composition may influence the kinetics of the Dox release. Higher doses of proteins in the medium may slower the kinetics due to their higher binding affinity. More hydrophobic composition of the culture media may enhance the Dox solubility. | ||
| Fig. 9 Cell viability MTT assay of Hela cells (A) and Insulinoma cells (B) treated with various nanogels measured after 48 h. Cells were cultured in vitro with free Dox and Dox loaded to PNIPA-co-AAc-, PNIPA-co-AAc-oligo1-2- and PNIPA-co-AAc-oligo1-2-3 nanogels. Inset: cell viability MTT assay recorded for blank nanogel carriers. | ||
We compared the cell viability results obtained from the fluorescence data with those from the UV-Vis data. The results were very similar. The mean differences obtained for both kinds of experiments done at 37 and 45 °C did not exceed 5 and 6%, respectively. Thus, we concluded that the efficiency of Dox release was not significantly affected by the possible quenching-of-fluorescence effect after internalization of the nanogels to the cells.
Cellular uptake of free Dox and Dox from particular nanogels was quantified after various treatment times of two cell lines, from 1 to 96 h. The concentration of Dox in treatment solution, including the unbound form and Dox incorporated into PNIPA-co-AAc-, PNIPA-co-AAc-oligo1-2-, and PNIPA-co-AAc-oligo1-2-3 NPs, was 10 μg mL−1. Fluorescence intensity of Dox, in the concentration range from 0 to 15 μg mL−1, was reported as linear vs. concentration.62 A comparison of fluorescence intensity of 1 μg mL−1 Dox dissolved in PBS, intercalated into dissolved in PBS oligo1-2-3 hybrid (c = 3.81 μmol mL−1), and incorporated into particular nanogels (c = 1.93 μmol mL−1 for or 3.81 μmol mL−1) is presented in Fig. 10. As it is seen in Fig. 10A, Dox fluorescence is dramatically quenched upon its intercalation into dsDNA.63 Our studies indicate that every fourth DNA base pair is occupied by one Dox molecule. Since in addition to intercalation the electrostatic interactions with the Dox tail also take place, the number of occupied base pairs is from two up to six.62
 | ||
| Fig. 10 Comparison of Dox fluorescence intensity determined for: (A) Dox dissolved in PBS and Dox intercalated into dsDNA dissolved in PBS, (B) Dox incorporated into particular nanogels. | ||
The kinetics of Dox intracellular accumulation in Hela and Insulinoma cells is presented in Fig. 11A–D. In all experiments the concentration of free Dox and Dox introduced into nanogels equaled 10 μg mL−1. Fig. 11A illustrates the intracellular accumulation of free Dox in Hela and Insulinoma cells. The fluorescence intensity is significantly lower for Insulinoma cells than Hela cells after 24 h and that decrease remained for further 60 h of incubation with free Dox. The decrease in Dox incubation in Insulinoma cells could be the effect of the increased detoxification capacity the drug resistance mechanisms are based on.62
 | ||
| Fig. 11 Cellular uptake of: (A) free Dox and (B) Dox released from PNIPA-co-AAc- (black), PNIPA-co-AAc-1-2- (red) and PNIPA-co-AAc-1-2-3 (blue) NPs, by Hela and Insulinoma cells. Comparison of accumulation of free Dox and Dox released from particular nanogels in Hela cells (C) and Insulinoma cells (D) after 96 h of treatment. | ||
Fig. 11B presents fluorescence intensities recorded for Hela and Insulinoma cells after releasing and absorbing of Doxorubicin from particular nanogels during 96 h of the cells treatment. Generally, for the nanogels with oligonucleotides we noticed lower fluorescence intensities in comparison to free Dox for both kinds of cancer cell lines. However, the positive result was that the fluorescence intensity of Dox released from PNIPA-co-AAc-oligo1-2-3 NPs increased linearly in time for both types of cancer cells. The fluorescence results clearly indicated that Dox can be efficiently delivered to the cells. The mean fluorescence intensities recorded for particular nanogels and free Dox accumulated after 96 h of the treatment of Hela and Insulinoma cells are presented in Fig. 11C and D, respectively. For both types of cancer cell lines, the mean fluorescence of free Dox is from 30% to 50% higher than that from particular nanogels.
4. Conclusions
In conclusion, for the first time a unique type of nanosized drug delivery system, based on PNIPA-co-AAc nanogels with covalent modification of the network with two specific oligonucleotides designed for the three-strand hybridization with the third oligonucleotide, was developed. The physicochemical parameters of synthesized novel nanogels: size and zeta potential promoted penetration of the cancer tissue. The presented nanogels appeared to be of good biocompatibility towards selected cancer cell lines. The synthesized PNIPA-co-AAc-oligo1-2-3 nanogels possessed the ability of improved storing, by intercalation to double stranded DNA, of selected anticancer drug doxorubicin (Dox). The ability to store and release of Dox, and the physiochemical parameters of novel PNIPA-co-AAc-oligo1-2-3 nanogels were compared with PNIPA-co-AAc- and PNIPA-co-AAc-oligo1-2 NPs, where oligo1 and oligo2 strands were non-complementary and therefore could not hybridize. The unique aspect of storing Dox by intercalation to a three-strand oligonucleotide hybrid attached to nanohydrogel net allowed us to design a drug delivery system with the prolonged time of drug release and therefore prolonged effective therapeutic action. Moreover, the selected type of Dox accumulation allowed us to ensure that the active form of the drug was well kept inside the composite network. In consequence, the possible toxic action of Dox in external physiological environment could be limited. We noticed that the mechanism of the Dox release from the nanogels at selected temperatures was a result of two reversible processes: oligo1-2-3 hybrid conformational change and shrinking of the hydrogel. Structural changes of PNIPA-co-AAc-oligo1-2-3 NPs resulted in their size- and zeta potential decrease. Finally, the synthesized nanogels worked well in in vitro experiments with Hela and Insulinoma cancer cells. Further investigations of developed multiple-factor nanobiomaterials with other types of intercalator drugs as the controlled delivery systems are in progress. Introduction of aptamers into the nanogels is planned. The results of this research may be also useful in construction of a specific DNA biosensor.Acknowledgements
Support of this work by grant no. 2012/05/D/ST5/03464 from the National Science Center of Poland is thankfully acknowledged.References
- H. Zhang, Y. Zhai, J. Wang and G. Zhai, Mater. Sci. Eng., C, 2016, 60, 560–568 CrossRef CAS PubMed.
- A. K. Hauser, R. J. Wydra, N. A. Stocke, K. W. Anderson and J. Z. Hilt, J. Controlled Release, 2015, 219, 76–94 CrossRef CAS PubMed.
- M. M. Perez-Madrigal, E. Armelin, J. Puiggali and C. Aleman, J. Mater. Chem. B, 2015, 3, 5904–5932 RSC.
- S. Mura, D. Trung Bui, P. Couvreur and J. Nicolas, J. Controlled Release, 2015, 208, 25–41 CrossRef CAS PubMed.
- X. Zeng, R. Morgenstern and A. M. Nystrom, Biomaterials, 2014, 35, 1227–1239 CrossRef CAS PubMed.
- S. Juodkazis, S. Mukai, R. Wakaki, A. Yamaguchi, S. Matsuo and H. Misawa, Nature, 2000, 408, 178–181 CrossRef CAS PubMed.
- G. Feng, H. Chen, J. Li, Q. Huang, M. J. Gupte, H. Liu, Y. Song and Z. Ge, Biomaterials, 2015, 52, 1–13 CrossRef CAS PubMed.
- J. K. Oh, D. J. Siegwart, H. I. Lee, G. Sherwood, L. Peteanu, J. O. Hollinger, K. Kataoka and K. Matyjaszewski, J. Am. Chem. Soc., 2007, 129, 5939–5945 CrossRef CAS PubMed.
- M. K. Pastuszka and J. A. MacKay, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2016, 8, 123–138 CrossRef CAS PubMed.
- Z. Shen, H. Wu, S. Yang, X. Ma, Z. Li, M. Tan and A. Wu, Biomaterials, 2015, 70, 1–11 CrossRef CAS PubMed.
- N. Wiradharma, Y. Zhang, S. Venkataraman, J. L. Hedrick and Y. Y. Yang, Nano Today, 2009, 4, 302–317 CrossRef CAS.
- M. Elsabahy and K. L. Wooley, Chem. Soc. Rev., 2012, 41, 2545–2561 RSC.
- W. Chen, K. Achazi, B. Schade and E. Haag, J. Controlled Release, 2015, 205, 15–24 CrossRef CAS PubMed.
- Z. Wu, X. Zhang, H. Guo, C. Lib and D. Yua, J. Mater. Chem., 2012, 22, 22788–22796 RSC.
- E. Molina, J. Warnant, M. Mathonnat, M. Bathfield, M. In, D. Laurencin, C. Jerome, P. Lacroix-Desmazes, N. Marcotte and C. Gerardin, Langmuir, 2015, 21, 12839–12844 CrossRef PubMed.
- Y. Tian, S. Bian and W. Yang, Polym. Chem., 2016, 7, 1913–1921 RSC.
- T. H. Senanayake, S. Gorantla, E. Makarov, Y. Lu, G. Warren and V. S. Vinogradov, Mol. Pharmaceutics, 2015, 12, 4226–4236 CrossRef CAS PubMed.
- A. M. Nowicka, E. Zabost, M. Donten, Z. Mazerska and Z. Stojek, Anal. Bioanal. Chem., 2007, 389, 1931–1940 CrossRef CAS PubMed.
- E. Zabost, W. Liwinska, M. Karbarz, E. Kurek, M. Lyp, M. Donten and Z. Stojek, Bioelectrochemistry, 2007, 109, 1–8 CrossRef PubMed.
- E. Zabost, A. M. Nowicka, Z. Mazerska and Z. Stojek, Phys. Chem. Chem. Phys., 2012, 14, 3408–3413 RSC.
- H. Yang, H. Liu, H. Kang and W. Tan, J Am Chem. Soc., 2008, 130, 6320–6321 CrossRef CAS PubMed.
- L. Peng, C. Wu, M. You, D. Han, Y. Chen, T. Fu, M. Ye and W. Tan, Chem. Sci., 2013, 4, 1928–1938 RSC.
- Y. Li, Y. Tseng, S. Kwon, L. d'Espaux, J. Bunch, P. McEuen and D. Luo, Nat. Mat., 2004, 3, 38–42 CrossRef CAS PubMed.
- W. Liao, C. Lu, R. Hartmann, F. Wang, Y. Sohn, W. Parak and I. Willner, ACS Nano, 2015, 9, 9078–9086 CrossRef CAS PubMed.
- H. Tang, A. Trondoli, G. Zhu, Y. Chen, Y. Chang, H. Liu, Y. Huang, X. Zhang and W. Tan, ACS Nano, 2011, 5, 5094–5099 CrossRef PubMed.
- A. Fujii, T. Maruyama, Y. Ohmukai, E. Kamio, T. Sotani and H. Matsuyama, Colloids Surf., A, 2010, 356, 126–133 CrossRef CAS.
- X. Zhang, D. Chabot, Y. Sultan, C. Monreal and M. DeRosa, ACS Appl. Mater. Interfaces, 2013, 5, 5500–5507 CAS.
- Z. Zhang, D. Balogh, F. Wang and I. Willner, J. Am. Chem. Soc., 2013, 135, 1934–1940 CrossRef CAS PubMed.
- Z. Wang, J. Xia, F. Cai, F. Zhang, M. Yang, S. Bi, R. Gui, Y. Li and Y. Xia, Colloids Surf., B, 2015, 134, 40–46 CrossRef CAS PubMed.
- L. Lee, A. Johnston and F. Caruso, Biomacromolecules, 2008, 9, 3070–3078 CrossRef CAS PubMed.
- Y. J. Wang, H. J. Xu, J. Wang, L. Ge and J. B. Zhu, J. Pharm. Sci., 2014, 103, 2012–2021 CrossRef CAS PubMed.
- D. E. Meyer, B. C. Shin and A. Chilkoti, J. Controlled Release, 2010, 74, 213–224 CrossRef.
- Y. Tiam, L. Bromberg, S. N. Lin, T. A. Hatton and K. C. Tam, J. Controlled Release, 2007, 121, 137–145 CrossRef PubMed.
- W. Schartl, Light scattering from polymer solutions and nanoparticle dispersions, Springer-Verlag, Berlin, Heidelberg, 2007 Search PubMed.
- I. Makra, P. Terejanszky and R. E. Gyurcsanyi, MethodsX, 2015, 2, 91–99 CrossRef PubMed.
- A. Kowalczyk, M. Fau, M. Karbarz, M. Donten and Z. Stojek, Biosens. Bioelectron., 2014, 54, 222–228 CrossRef CAS PubMed.
- M. Mackiewicz, K. Kaniewska, J. Romanski, E. Augustin, Z. Stojek and M. Karbarz, J. Mater. Chem. B, 2015, 3, 7262–7270 RSC.
- X. Hu, X. Hao, Y. Wu, J. Zhang, X. Zhang, P. Wang, G. Zou and X. Liang, J. Mater. Chem. B, 2013, 1, 1109–1118 RSC.
- I. Galaev and B. Mattiasson, Smart Polymers, Application in Biotechnology and Biomedicine, CRC Press, New York, 2nd edn, 2008 Search PubMed.
- W. Qi, B. Song, X. Lei, C. Wang and H. Fang, Biochemistry, 2011, 50, 9628–9632 CrossRef CAS PubMed.
- M. Hayat, Stains and cytochemical methods, Plenum Press. New York, 1993 Search PubMed.
- R. Ziółkowski, Ł. Górski, S. Oszwałdowski and E. Malinowska, Anal. Bioanal. Chem., 2012, 402, 2259–2266 CrossRef PubMed.
- T. Mizuhara, D. Moyano and V. Rotello, Nano Today, 2016, 11, 31–40 CrossRef CAS PubMed.
- H. Chien, Y. Gu, Y. Hu and Z. Quian, J. Pharm. Sci. Technol., 2007, 4, 303–313 Search PubMed.
- H. Mansur, C. Sadahira, A. Souza and A. Mansur, Mater. Sci. Eng., C, 2008, 28, 539–548 CrossRef CAS.
- J. Martens, C. Rogero, M. Calleja, D. Ramos, J. Martin-Gago, C. Briones and J. Tamayo, Nat. Nanotechnol., 2008, 3, 301–307 CrossRef PubMed.
- C. Chen, Z. Lai, G. Wang and C. Wu, Biosens. Bioelectron., 2016, 77, 603–608 CrossRef CAS PubMed.
- B. Zhu, M. Booth, P. Shepherd, A. Sheppard and J. Travas-Sejdic, Biosens. Bioelectron., 2015, 64, 74–80 CrossRef CAS PubMed.
- A. Lasia, in Modern aspects of electrochemistry modeling and numerical simulations, ed. M. Schlesinger, Springer, New York, 2009 Search PubMed.
- S. Grutzke, S. Abali, W. Schuhmann and M. Gebala, Electrochem. Commun., 2012, 19, 59–62 CrossRef.
- E. Zabost, W. Chmielowiec, T. Rapecki, M. Karbarz and Z. Stojek, Electrochem. Commun., 2014, 42, 50–54 CrossRef CAS.
- E. Zabost, A. M. Nowicka, M. Donten and Z. Stojek, Phys. Chem. Chem. Phys., 2009, 11, 8933–8938 RSC.
- X. Zeng, R. Morgernstern and A. Nystrom, Biomaterials, 2014, 35, 1227–1239 CrossRef CAS PubMed.
- A. Hervault, A. Dunn, M. Lim, C. Boyer, D. Mott, S. Maenosono and N. Thann, Nanoscale, 2016, 8, 12152–12161 RSC.
- C. Pérez-Arnaiz, N. Busto, J. M. Leal and B. García, J. Phys. Chem. B., 2014, 118, 1288–1295 CrossRef PubMed.
- J. D. McGhee and P. H. von Hippel, J. Mol. Biol., 1974, 86, 69–489 CrossRef.
- M. Airoldi, G. Barone, G. Gennaro, A. M. Giuliani and M. Giustini, Biochemistry, 2014, 53, 2197–2207 CrossRef CAS PubMed.
- P. Ritger and N. Peppas, J. Controlled Release, 1987, 5, 23–36 CrossRef CAS.
- C. Sanson, C. Schatz, J. Le Meins, A. Soum, J. Thevenot and E. Garanger, et al., J. Controlled Release, 2010, 147, 428–435 CrossRef CAS PubMed.
- T. Gan, Y. Guan and Y. Zhang, J. Mater. Chem., 2010, 20, 5937–5944 RSC.
- J. van Meerloo, G. J. Kaspers and J. Cloos, Cancer Cell Culture, 2011, 731, 237–245 CAS.
- P. Mohan and N. Rapoport, Mol. Pharm., 2010, 7, 959–1973 Search PubMed.
- H. T. Haj, M. Salerno, W. Priebe, H. Kozlowski and A. Garnier-Suillerot, Chem.–Biol. Interact., 2003, 145, 349–358 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra16592c |
| This journal is © The Royal Society of Chemistry 2016 |