
Transporter engineering and enzyme evolution for pyruvate production from d/l-alanine with a whole-cell biocatalyst expressing l-amino acid deaminase from Proteus mirabilis†
Abstract
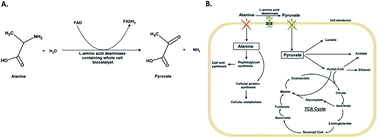
Pyruvate is an essential metabolite in the central metabolism of microbes, and it has been widely used in the food, pharmaceutical, and agrochemical industries. Both chemical and biological processes have been used for industrial pyruvate production. In this study, one-step pyruvate production from D/L-alanine with a whole-cell E. coli biocatalyst expressing L-amino acid deaminase (pm1) from Proteus mirabilis was investigated. Alanine uptake transporters (cycA, amaP) and a pyruvate uptake transporter (lldP) were knocked out to prevent substrate and product utilization by the biocatalyst. The pyruvate production titer from D/L-alanine increased from 1.14 g L−1 under control conditions to 5.38 g L−1 with the mutant whole-cell biocatalyst. Directed evolution was used to engineer pm1 and improve the catalytic activity with D/L-alanine. Three rounds of error-prone polymerase chain reaction generated the mutant pm1ep3, which showed improved affinity (6.76 mM for L-alanine) and catalytic efficiency (0.085 s−1 mM−1 and 0.027 s−1 mM−1 for L- and D-alanine, respectively). The final pyruvate titer was increased to 14.57 g L−1 and the conversion ratio was increased to 29.14% by using the engineered whole-cell biocatalyst containing the evolved pm1ep3.
 Please wait while we load your content...
Please wait while we load your content...

