DOI:
10.1039/C6RA16296G
(Paper)
RSC Adv., 2016,
6, 79228-79235
Effects of organosolv fractionation time on thermal and chemical properties of lignins†
Received
23rd June 2016
, Accepted 10th August 2016
First published on 10th August 2016
Abstract
Organosolv fractionation is a promising pathway to separate cellulosic biomass into high purity cellulose, hemicelluloses, and lignin. This work specifically investigates the properties of lignins isolated at specific time points as fractionation progressed, with the intent of correlating fractionation time with lignin purity, yield, thermal and chemical properties. Yellow poplar (Liriodendron tulipifera) was fractionated using a mixture of methyl isobutyl ketone, ethanol, and water with sulfuric acid as catalyst at 140 °C over a two-hour period. Aliquots of the liquor were collected by sampling every 15 min during the fractionation to generate a series of lignins. The results showed that with increased fractionation time, lignin purity improved from 90.3 to 94.6% and the glass transition temperature increased from 117 to 137 °C. The loss of aliphatic OH and increase of phenolic OH with fractionation time led to an increase in condensed structures and increased polydispersity at times greater than 90 min. Principal component analysis of Fourier transform infrared spectroscopic data confirmed the shift to higher purity and more condensed chemical structures with increasing fractionation time. Overall, this study demonstrates that thermal and chemical properties of lignin change with the organosolv fractionation time.
Introduction
Public, corporate, and policy makers globally recognize a need to find alternatives to fossil sources for carbon based fuels, chemicals, and materials to limit greenhouse gas emissions. Lignocellulosic biomass has come to the forefront as a viable alternative to replace much of our carbon demand for energy and products, while leaving a smaller environmental footprint than fossil equivalents. To enable the use of lignocellulosic based carbon, one must deconstruct biomass into its individual polymeric components, cellulose, hemicelluloses, and lignin, to enable their efficient conversion into desired products. Of the available commercial technologies, organosolv fractionation has proven to deliver separate streams of biomass polymers with high purity.1,2 The carbohydrate streams, cellulose and hemicelluloses, can be converted readily to products, such as fibers, fuels, and chemicals, via mature technologies developed over decades.
Lignin, unfortunately, has always been considered a waste or low value byproduct that is burned for fuel or landfilled instead of being used as a valued feedstock for chemicals and materials. The majority of commercially pulping technologies renders lignin infusible and contaminated with a host of inorganics, residual sugars, degradation products, and other organic impurities. A recent study compared the structure and composition of six industrial lignins from three major industrial pulping methods (Indulin AT Kraft, Portobind 1000 soda, Alcell, poplar, spruce, and wheat straw ethanol based organosolv lignin).3 It showed that the high purity organosolv lignin had the advantage of higher catalytic stability during the catalytic depolymerisation. More recently, a mixture of water/ethanol/methyl isobutyl ketone (MIBK) was proven to successfully fractionate biomass into three clean fractions: lignin, hemicellulose derived sugar fragments, and cellulose.4 This fractionation technology has proven to be an effective method for producing lignin at high yields and high in purity.5–7 Under the correct processing conditions, the organosolv fractionation process produces fusible lignin without the addition of other plasticizers or chemical modification, which can enhance its utility as a feedstock for carbon fibers, thermoplastics, or polymer blends.5,6,8 However, previous results demonstrate that fractionation conditions can produce lignin with a variety of thermal properties (i.e. glass transition temperature, Tg) that strongly correlate to the material chemistry.8 Several factors influenced this result, namely species, pulping solvent, conditions, and severity.
Various organosolv solvent mixtures have been proposed for lignin removal from biomass, such as ethanol/water,9,10 acetic acid/formic acid/water,11–13 acetone/water,14–16 and high boiling point solvent/water.17 A previous study using acetic acid organosolv pulping found no impact on lignin molecular weight and only slight changes to lignin hydroxyl functionality for lignin aliquots sampled at different times during processing.18 However, this process has relatively low yields (≤65%). Lignin chemistry from the kraft process is more dependent upon the processing time and conditions, where lignin is severely degraded at long processing times.19 There were a few attempts to investigate the effect of the reaction severity on the ethanol based organosolv lignin characteristics. Higher organosolv temperature reduced amount of residual carbohydrates in lignin.9 With the increase of combined reaction severity, lignin showed a decrease in molecular weight, more severe dehydration, and condensation reactions.20 For the organosolv fractionation process, we hypothesize that for a given set of conditions, lignin of varying chemistry will be released at different times in the process as the kinetics to cleave certain bonds present in the biomass differ. This will offer a potential method to separate lignin into fractions with unique molecular weights and chemistries for specific applications, such as low molecular weight for chemical feedstocks and higher molecular weights for polymers. Therefore, our objective was to investigate the thermal and chemical properties of the lignins as fractionation progresses to discern differences and implications on their use.
In this study, organosolv fractionation was performed on yellow poplar over a two-hour reaction period. The black liquor was collected every 15 min during the fractionation to generate a series of lignins. In order to investigate the chemical and thermal properties of the generated lignins, the chemical composition, C and H content, molecular weight, glass transition temperature (Tg), and lignin decomposition temperature were measured. In addition, Fourier transform infrared (FT-IR) and nuclear magnetic resonance (NMR) spectroscopy were used to explore the chemical structures and functionalities of the generated lignins.
Experimental
Materials
Pulp grade yellow poplar (Liriodendron tulipifera) chips (dimension: 2 × 2 × 0.05 cm), free of bark, were purchased from Oak Ridge Hardwoods. The chips were air-dried to a moisture content of 9%. The lignin content of the material chips was determined to be 21% (dry weight basis). All chemicals in this study were purchased from Fisher Scientific and used as received.
Organosolv fractionation
The biomass was fractionated according to an established method.4 Briefly, yellow poplar chips (∼660 g) were loaded in a flow-through reactor. The reactor was sealed and the chips were impregnated with a 16/34/50 wt% solution of MIBK, ethanol, and water, respectively with sufficient sulfuric acid to make a 0.05 M solution. The reactor was electrically heated to 140 °C, and the solution was then pumped through the reactor for 120 minutes at a rate sufficient to generate an ultimate solvent to biomass ratio of 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.21 After the reactor temperature reached 140 °C, all the liquor generated during the first 30 min was collected. Subsequently, the liquor was collected every 15 min with the last collection time at 120 min. A series of seven liquor samples was collected in total. After 120 min, the liquor remaining in the reactor was collected to give a final fraction. A control run was also performed using the same reaction conditions (140 °C and 120 min) except that the liquor was collected as one batch after 120 min. Lignin was isolated from each liquor fraction by adding solid sodium chloride (15 g per 100 mL) in a separatory funnel to induce a phase separation between the aqueous and organic layers.21 After the removal of the aqueous phase, the organic phase was washed twice with 30% (v/v) deionized water to remove additional ethanol and sugars. The ethanol in the combined aqueous phase was removed using a rotary evaporator to give an aqueous solution with a lignin precipitate that was isolated by filtration, air dried, and blended with the organic phase. The organic solvent was removed from the combined fraction on a rotary evaporator and the resulting lignin was triturated five times with 50 mL of diethyl ether and water washed by stirring in a beaker (solid to liquid ratio of 1:20) overnight. Each lignin fraction was filtered, dried overnight in a vacuum oven at 80 °C, and weighed.
:1.21 After the reactor temperature reached 140 °C, all the liquor generated during the first 30 min was collected. Subsequently, the liquor was collected every 15 min with the last collection time at 120 min. A series of seven liquor samples was collected in total. After 120 min, the liquor remaining in the reactor was collected to give a final fraction. A control run was also performed using the same reaction conditions (140 °C and 120 min) except that the liquor was collected as one batch after 120 min. Lignin was isolated from each liquor fraction by adding solid sodium chloride (15 g per 100 mL) in a separatory funnel to induce a phase separation between the aqueous and organic layers.21 After the removal of the aqueous phase, the organic phase was washed twice with 30% (v/v) deionized water to remove additional ethanol and sugars. The ethanol in the combined aqueous phase was removed using a rotary evaporator to give an aqueous solution with a lignin precipitate that was isolated by filtration, air dried, and blended with the organic phase. The organic solvent was removed from the combined fraction on a rotary evaporator and the resulting lignin was triturated five times with 50 mL of diethyl ether and water washed by stirring in a beaker (solid to liquid ratio of 1:20) overnight. Each lignin fraction was filtered, dried overnight in a vacuum oven at 80 °C, and weighed.
Chemical composition and elemental analysis
Lignin purity, carbohydrates, and ash content were determined using standard procedures (NREL/TP-510-42618 and NREL/TP-510-42622). In brief, approximately 300 mg lignin sample went through a two-step acid hydrolysis and separated into soluble and insoluble fractions by filtration. The acid soluble lignin content was measured using a UV-Vis spectrophotometer (UV absorption at 205 nm, absorption coefficient of 110 L g−1 cm−1) (Thermo Scientific GENESYS 10S). The carbohydrates (hydrolyzed to mono-sugars) content was quantified using a high performance liquid chromatograph (Perkin Elmer 200 series) equipped with a refractive index detector and an Aminex HPX-87P column (Bio-Rad). The acid insoluble lignin was calculated by gravimetric method after ashing the insoluble fraction at 575 °C for 24 h in a muffle furnace (Fisher Scientific). The ash content of each lignin was determined gravimetrically after combustion of the material at 575 °C for 24 h in a muffle furnace (Fisher Scientific). Elemental analysis of the lignin samples was performed using a CHN analyzer (Perkin Elmer Series II 2400 CHN S/O). All measurements were performed in triplicate.
Molecular weight
The weight average molecular weight (Mw), number average molecular weight (Mn), and polydispersity index (Mw/Mn, PDI) of the lignins were determined by gel permeation chromatography (GPC). Approximately 50 mg of a lignin sample were acetylated with 1 mL of pyridine/acetic anhydride (1:1, v/v) and stirred for 24 h at room temperature. The reaction was mixed with ethanol (25 mL), stirred for 30 min, and the reaction solvent removed by rotary evaporation. The addition of ethanol and rotary evaporation of the solvent were repeated several times until the pyridine and acetic anhydride were completely removed. Subsequently, the acetylated lignin was dissolved in chloroform and added to anhydrous diethyl ether. The lignin was then recovered by centrifugation and further washed three times with 100 mL of diethyl ether. The recovered lignin was dried in a 40 °C vacuum oven for 24 h.20,22 The acetylated lignin was dissolved in tetrahydrofuran (THF), filtered through a 0.45 μm filter, and immediately analyzed using a size exclusion chromatography system (Tosoh ECO SEC) equipped with a UV detector (265 nm). The analysis was carried out with THF as the eluent (0.35 mL min−1) on a Tosoh TSK gel Super Multipore HZ-M column (4.6 × 150 mm, 4 μm packing) preceded by a TSK gel Super Multipore HZ-M guard column and calibrated against polystyrene.
31P NMR spectroscopy
31P NMR was performed after derivatization of the lignin samples with 2-chloro-4,4,5,5-tetramethyl-1,3,2-dioxaphospholane (TMDP). Cyclohexanol and chromium(III) acetylacetonate were used as an internal standard and a relaxation agent, respectively.23,24 The quantitative 31P NMR spectra were recorded using a Varian 400-NMR spectrometer at frequency of 162 MHz using a 90° pulse angle, 25 s pulse delay, and 256 transients at room temperature.24,25
Fourier transform infrared spectroscopy (FTIR)
A small amount of lignin was placed on a diamond attenuated total reflectance cell of a Perkin-Elmer (Waltham, MA) Spectrum One FTIR Spectrometer. Spectra were collected from 4000 to 600 cm−1 in absorbance mode with eight scans per spectrum at 4 cm−1 resolution. Ten individual spectra were collected for each lignin sample.
Thermal properties of the lignins
Differential scanning calorimetry (DSC) was performed on a Perkin Elmer Diamond DSC (Shelton, CT) under nitrogen atmosphere. Approximately 2 mg of lignin were hermetically sealed in an aluminium pan. The lignin was first heated from 25 to 200 °C at a rate of 100 °C min−1 to minimizing the possible crosslinking or other kinetic processes during heating, annealed to eliminate any stored thermal history within the polymer's glassy state, re-cooled to 25 °C, and then reheated to 200 °C at the same heating rate. The high heating rate enhances weak transitions, such as those observed in lignin and other natural polymers.26 The analysis was performed in triplicate.
Thermogravimetric analysis (TGA) was carried out using a Perkin Elmer Pyris 1 TGA (Shelton, CT). Approximately 5–7 mg of lignin were placed in a platinum pan, heated under nitrogen at a rate of 10 °C min−1 from 20 to 105 °C, and held for 10 min to remove moisture. The sample was then heated to 950 °C at the same heating rate. The average main decomposition temperature and weight yield at 900 °C were calculated based on two replicates.
Statistical analysis
In order to assess the chemical differences in the FTIR spectra, principal component analysis (PCA) was performed over the IR range of 4000 to 600 cm−1. PCA was performed using the Unscrambler software package (ver.9.0; CAMO Process AS, Norway). Prior to the analysis, the spectra were transformed by mean normalization. Full multiplicative scatter correction (MSC) was applied to compensate for scatter and baseline effects in the data.
In order to detect hidden relationships within and/or between the thermal and chemical properties of the lignins isolated at different fractionation times, a Pearson's correlation analysis was carried out using SAS 9.3 (Cary, NC). Correlation coefficients and p-values were calculated.
Results and discussion
Isolated lignin yield and chemical composition
Yield and purity data of lignins fractionated at different times are presented in Fig. 1 and Table 1, respectively. The lignin yields are adjusted based on lignin purity. The seven isolated lignins from the fractionation added up to 111.9 g, which corresponded to 80.5% of the total lignin present in the biomass. The parallel control fractionation afforded 119.6 g of lignin, which was 86.0% of that present in the yellow poplar. To compare, a previous hybrid poplar ethanol based organosolv reaction (50% ethanol, 180 °C, 60 min, and 1.25% H2SO4) had a precipitated lignin yield at 74% of the Klason lignin in untreated wood.27 In our study, the change in lignin yield with respect to the reaction time was closely monitored. The lignin yield increased from 13.1% at 30 min to 15.7% at 45 min. As the fractionation time increased to 120 min, the yield of lignin decreased gradually to 4.0%. The last liquor collection (final draining) from the reactor at completion of the fractionation contained 9.0% of the total lignin. More than 70% of the lignin present in the biomass were extracted within 90 min of the fractionation and 9.7% of the total lignin were obtained during the last 30 min of the reaction. The decrease of lignin yield with the fractionation time could be due to a decrease in available lignin present in the material.
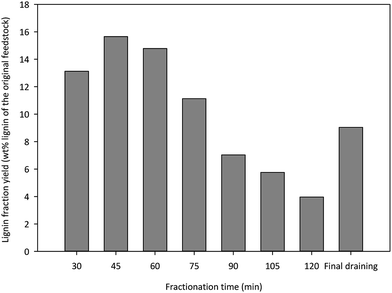 |
| | Fig. 1 The lignin fraction yield (wt% lignin of the original feedstock) at different fractionation time periods. | |
Table 1 The lignin chemical composition and elemental analysis at different fractionation time periods (mean ± standard deviation)
| Fractionation time (min) |
Chemical composition (wt% on dry basis) |
Elemental analysis (%) |
| Ash |
Glucan |
Xylan + mannan |
Acid insoluble lignin |
Acid soluble lignin |
Lignin purity |
C |
H |
| ND: not detected. |
| 30 |
0.1 ± 0.0 |
0.9 ± 0.0 |
3.6 ± 0.0 |
87.3 ± 0.3 |
3.1 ± 0.2 |
90.3 ± 0.0 |
61.8 ± 0.5 |
6.2 ± 0.1 |
| 45 |
0.1 ± 0.0 |
NDa |
2.7 ± 0.0 |
89.5 ± 0.6 |
2.0 ± 0.1 |
91.5 ± 0.7 |
63.0 ± 0.3 |
6.0 ± 0.0 |
| 60 |
0.1 ± 0.0 |
ND |
2.6 ± 0.0 |
91.2 ± 0.6 |
2.2 ± 0.1 |
93.8 ± 0.3 |
63.2 ± 0.2 |
6.0 ± 0.1 |
| 75 |
0.1 ± 0.0 |
ND |
2.5 ± 0.0 |
92.0 ± 0.5 |
1.8 ± 0.2 |
93.8 ± 0.3 |
63.7 ± 0.2 |
5.9 ± 0.1 |
| 90 |
0.1 ± 0.0 |
ND |
2.4 ± 0.0 |
92.4 ± 0.4 |
1.9 ± 0.1 |
94.2 ± 0.4 |
63.6 ± 0.7 |
5.7 ± 0.3 |
| 105 |
0.1 ± 0.0 |
ND |
2.3 ± 0.0 |
92.4 ± 0.3 |
2.0 ± 0.1 |
94.4 ± 0.2 |
63.6 ± 0.1 |
5.7 ± 0.0 |
| 120 |
0.1 ± 0.0 |
ND |
2.1 ± 0.0 |
92.9 ± 0.2 |
1.7 ± 0.0 |
94.6 ± 0.2 |
63.3 ± 0.5 |
5.7 ± 0.1 |
| Control |
0.1 ± 0.0 |
ND |
2.5 ± 0.1 |
90.7 ± 0.5 |
1.7 ± 0.1 |
92.4 ± 0.5 |
62.9 ± 0.1 |
6.1 ± 0.1 |
As the organosolv fractionation progressed, lignin purity increased, ash content remained constant at 0.1%, and carbohydrates content decreased. Lignin purity was calculated by combining the acid insoluble and acid soluble lignin fractions. The lowest purity (90.3%) was obtained at the shortest fractionation time (30 min), and then the purity augmented with fractionation time and plateaued at 94% after 90 minutes. The purity of the control lignin was 92.4%, which was slightly higher than the lignin fraction generated at 30 min, similar to the 45 min lignin, but lower as compared to fractions generated at and after 60 min. The highest acid soluble lignin content (3.1%) was obtained at the shortest time fraction (30 min) and the lowest content (1.7%) was obtained at the longest time (120 min). The opposite was observed for acid insoluble lignin, with the lowest amount (87.3%) at 30 min and the highest value (92.9%) at 120 min. The major impurity in this series of lignin was composed of xylan and mannan. It ranged from 2.1 to 3.6%, decreasing with fractionation time. Glucan was only detected in the 30 min sample (0.9%). The lignin collected at shorter fractionation time (lower reaction severity) had higher carbohydrate content. This agrees with a previous ethanol based organosolv study.28 The control lignin exhibited xylan and mannan content of 2.5% and no glucan.
Elemental analysis of the lignins
As expected, the 30 min sample, which had the highest amount of carbohydrates, also had the lowest carbon (C) content (61.8%) and the highest hydrogen (H) content (6.2%) as shown by the elemental analysis results of the lignin fractions in Table 1. The C content of lignins produced at shorter fractionation times (30 and 45 min) was slightly lower compared to that of the lignins fractionated at ≥60 min (63.2%). This finding validated the chemical composition data that lignin isolated at the beginning of the fractionation possessed a higher amount of organic impurities. The higher C content in lignin also would be a beneficial factor when lignin is used as the precursor for the carbon fiber production.29
Molecular weight distribution
The number-average (Mn) and weight-average molecular weights (Mw), and polydispersity index (PDI) for all lignin samples are summarized in Table 2. Both Mn and Mw generally decreased with fractionation time. Mn ranged from 900 to 2000 g mol−1 while Mw ranged from 5700 to 8300 g mol−1, giving PDI values varied between 3.6 and 6.6. Between fractionation times of 30 and 105 min, PDI increased from 4.6 to 6.6. However, a decrease (4.1) was observed at the longest time (120 min). Interestingly, the lowest PDI value (3.8) was observed for the lignin obtained from the control fractionation. This difference between the control and the lignins fractionated by different times could be due to possible additional chemical reactions that took place between the early and later extracted lignin. The Mw of the 30 min lignin was the highest among all fractions, possibly due to minimal fragmentation and side reactions of the lignins at the early stages of the organosolv reaction.30 Leskinen et al.31 compared the lignin isolated by various refining methods and concluded that ethanol acid catalyzed reaction generated lignins with the lowest Mw and PDI among various methods.
Table 2 The number average molecular weight (Mn), weight average molecular weight (Mw), and polydispersity index (PDI) of lignins at different fractionation time periods
| Fractionation time (min) |
Mn (g mol−1) |
Mw (g mol−1) |
PDI |
| 30 |
1800 |
8300 |
4.6 |
| 45 |
2000 |
8300 |
4.2 |
| 60 |
1400 |
6000 |
4.3 |
| 75 |
1800 |
6900 |
3.8 |
| 90 |
1300 |
7100 |
5.5 |
| 105 |
900 |
5900 |
6.6 |
| 120 |
1400 |
5700 |
4.1 |
| Control |
1900 |
6900 |
3.6 |
31P NMR spectroscopy
Since hydroxyl groups are one of the most important functionalities that affect the chemical and physical properties of lignin,24 31P NMR was used to quantify the different hydroxyl groups present in the lignins and assess their reactivity at different fractionation times. As presented in Table 3, the dominant hydroxyl group in the 30 min lignin was aliphatic OH groups (3.41 mmol g−1). At longer fractionation times, aliphatic OH decreased to 2.05 mmol g−1 (120 min). Phenolic OH concentration in the lignin fractions augmented from 1.69 to 3.52 mmol g−1 with fractionation time. The loss of aliphatic OH and increase of phenolic OH with increasing fractionation time presumably resulted from the acid catalyzed elimination of aliphatic OH, and concomitant formation of new phenolic OH via β-aryl ether linkages.32 Similar results were observed by El Hage et al.20 while comparing the OH concentration of milled wood lignin and organosolv lignins with different reaction severities from Miscantus. Milled wood lignin had lower phenolic OH and higher aliphatic OH concentration compared to organosolv lignins. With the organosolv reaction severity increased, a strong increase in the phenolic OH (from 2.34 to 3.93 mmol g−1) and decrease in aliphatic OH (from 3.11 to 1.26 mmol g−1) were found in organosolv lignins.20 Increase in p-hydroxyphenyl and syringyl OH from none to 0.07 and 0.98 to 2.51 mmol g−1, respectively, was observed with increasing fractionation time as a result from cleavage of ether bonds.33 Carboxylic acid OH content slightly reduced from 0.07 at 30 min to 0.03 mmol g−1 at 120 min. The total OH of the fractions before 45 min was constant at 5.17 mmol g−1 while at the end of fractionation total OH reached 5.64 mmol g−1. The increase in the total OH showed elimination of aliphatic OH. Organosolv lignin with high aliphatic OH concentration has been proven to be polyols for manufacturing polyurethanes34 and polyurethane foams,35 while lignin with high phenolic and low aliphatic OH content showed improved antioxidant properties for lignin–polypropylene blend.36 Therefore, it is possible to tailor lignin with different functionalities for various applications by controlling the organosolv reaction time.
Table 3 Hydroxyl group contents of lignin samples (mmol g−1) calculated by 31P NMRa
| Fractionation time (min) |
COOH (mmol g−1) |
Phenolic OH (mmol g−1) |
Total phenolic OH (mmol g−1) |
Aliphatic OH (mmol g−1) |
Total OH (mmol g−1) |
| Cond. |
H |
G |
S |
| COOH: carboxylic acid; Cond.: condensed; H: p-hydroxyphenyl; G: guaiacyl; S: syringyl. |
| 30 |
0.07 |
0.18 |
0.00 |
0.53 |
0.98 |
1.69 |
3.41 |
5.17 |
| 45 |
0.04 |
0.22 |
0.03 |
0.52 |
1.56 |
2.33 |
2.80 |
5.17 |
| 60 |
0.05 |
0.41 |
0.03 |
0.63 |
1.70 |
2.77 |
2.70 |
5.52 |
| 75 |
0.05 |
0.29 |
0.04 |
0.62 |
2.07 |
3.02 |
2.52 |
5.59 |
| 90 |
0.06 |
0.41 |
0.06 |
0.58 |
1.78 |
2.82 |
2.58 |
5.46 |
| 105 |
0.05 |
0.33 |
0.05 |
0.71 |
2.15 |
3.24 |
2.27 |
5.56 |
| 120 |
0.07 |
0.31 |
0.07 |
0.65 |
2.51 |
3.52 |
2.05 |
5.64 |
| Control |
0.03 |
0.26 |
0.03 |
0.67 |
1.81 |
2.71 |
2.89 |
5.63 |
Fourier transform infrared spectroscopy (FTIR)
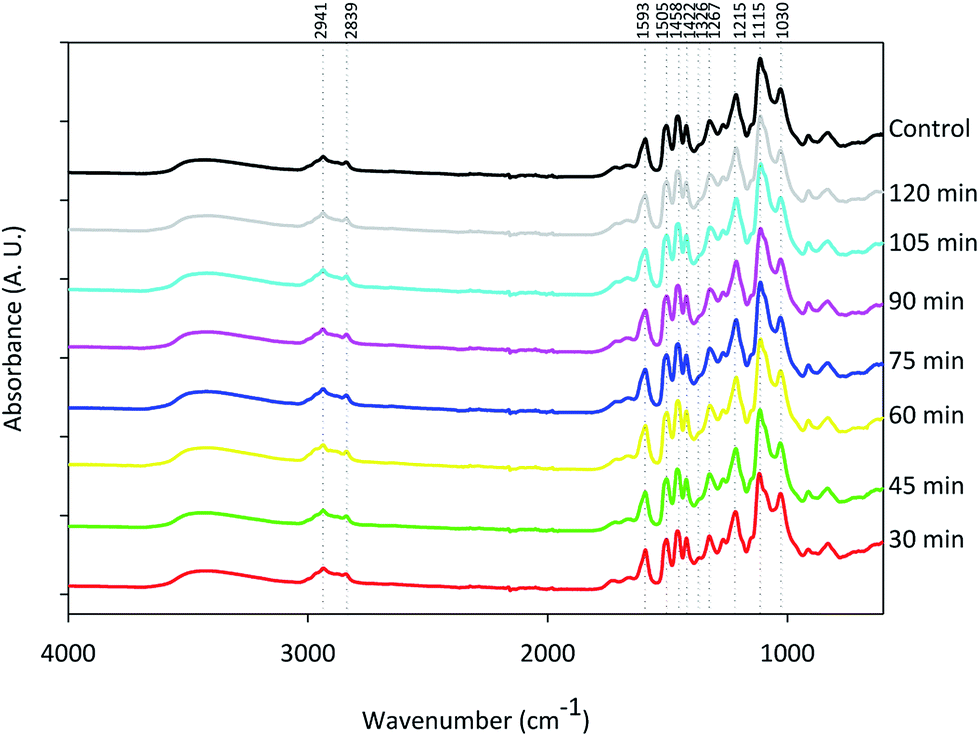
The type of functional groups presented in the isolated lignins in function of time was investigated by FTIR. Averaged FTIR spectra of all the lignin samples in the range 4000 to 600 cm−1 are shown in Fig. 2 with major peaks labeled and assigned in Table 4. PCA, a statistical method that illustrates correlation among multivariable data and determines the primary and secondary differences in large and complex dataset, was performed on these spectra. Each principal component (PC) describes the variability of the variables in the data with PC1 accounting for the largest possible variance, PC2 the second largest variance in the data, and so on. The scores plot showing the separation by grouping similar samples and helping to interpret the relationships among samples is provided in Fig. 3(a) with PC1 accounting for 58% and PC2 for 32% of the total spectral variance, respectively. The loadings plot indicating the relationship between the original variables (wavenumber) and PCs is shown in Fig. 3(b). Lignins isolated at different reaction times separated along PC1. Samples obtained at 30, 45, 60 min and under the control conditions located in the positive quadrant of PC1, whereas lignin samples obtained under longer times closely clustered together in the negative quadrant of PC1. Moreover, as reaction time increased, the fractionated lignins shifted from the positive to the negative axis of PC1, indicating that a relationship existed between fractionation time and the corresponding lignin chemical structure. Once again, the 30 min lignin differed the most from the other fractions and was located at the greatest distance from the other lignins along the PC1 axis. Fig. 3(b) identified the major infrared bands responsible for this separation. The strong positive bands at 1031 cm−1 (C–H deformation in guaiacyl with C–O deformation in the primary alcohol vibration) and 1123 cm−1 (C–O–C asymmetric stretching vibration), and the location for 30, 45, and 60 min lignins were consistent with the NMR results and confirmed that these lignins contained more aliphatic alcohol functional groups or C–H bending in plane in guaiacyl units than the longer time samples. In addition, these shorter time lignins contained lower intensity bands at 1195 cm−1 (probably arising from O–H bending vibration) and 1303 cm−1 (CH2 rocking vibration). The minor negative band at 1650 cm−1 and positive band at 1743 cm−1 (Fig. 3(b)) indicated a change in lignin reactivity as well: less conjugated carbonyl groups and more ester linkages in the short time lignin samples. The broad positive peak at 3451 cm−1 corresponding to hydroxyl stretching indicated the higher aliphatic OH groups content in the lignin fractionated at shorter times.
 |
| | Fig. 2 Averaged FTIR spectra for lignin samples. | |
Table 4 Assignment of FTIR bands37–39
| Bands (cm−1) |
Assignment |
| 2941 |
C–H stretch in OCH3 |
| 2838 |
C–H stretch in OCH3 |
| 1590 |
Aromatic skeletal vibration plus C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretch O stretch |
| 1505 |
Skeletal vibration |
| 1458 |
C–H deformation methyl and methylene |
| 1422 |
Aromatic ring stretching with in plane C–H deformation |
| 1326 |
C–O stretch of syringyl ring |
| 1267 |
C–O stretch of guaiacyl ring |
| 1215 |
C–C, C–O, and CO stretching of guaiacyl unit |
| 1115 |
Aromatic C–H deformation of syringyl unit |
| 1030 |
C–H bending in plane in guaiacyl units and C–O stretch of primary alcohol |
 |
| | Fig. 3 (a) FTIR principal component analysis (PCA) scores plot of lignin with different fractionation time and control lignin. (b) Corresponding PC1 loadings plot of the FTIR of lignin with different fractionation time and control lignin. | |
Thermal properties
The glass transition temperature (Tg) is an essential parameter for lignin processing in various applications; for example, when using lignin as carbon fiber precursors, the Tg has a strong impact on the melting-processing or stabilization temperature prior to carbonization.40 The Tg of each lignin fraction is given in Table 5. The 120 min lignin had the highest Tg at 137 °C whereas the 30 min sample had the lowest at 117 °C. The control lignin had an intermediate Tg of 122 °C. Lignin glass transition temperature could be influenced by several molecular factors such as crosslinking, intermolecular forces, and molecular weight.41 The increase of the Tgs with fractionation time can be indicative of a more condensed, C–C, structure were formed.42 More significantly, the increased Tg usually indicates a higher aliphatic OH content as these groups form stronger hydrogen bonds. This seems to contradict the results obtained from 31P NMR analysis. However, it is the increase in phenolic OH content with time that suggests the formation of more C–C bonds from severed β-O-4 linkages. This increase in condensed structures and total OH content has the most profound impact on increasing Tg with fractionation time, not molecular weight.
Table 5 Glass transition temperature (Tg) of lignins isolated at different time measured by differential scanning calorimetry (with standard deviation of 0.5 °C for Tg), and the % yield at 900 °C and lignin decomposition temperature by thermogravimetric analysis
| Fractionation time (min) |
Tg (°C) |
% yield at 900 °C |
Main decomposition temperature (°C) |
| 30 |
117 |
31.0 |
369 |
| 45 |
127 |
33.1 |
379 |
| 60 |
124 |
33.6 |
380 |
| 75 |
126 |
35.0 |
382 |
| 90 |
134 |
36.4 |
382 |
| 105 |
133 |
36.8 |
375 |
| 120 |
137 |
37.7 |
384 |
| Control |
122 |
33.1 |
376 |
The thermal stability of the lignin samples as indicated by the main decomposition temperature and the weight loss at 900 °C is summarized in Table 5. Compared to lignins produced at longer fractionation time, 30 min lignin had the lowest char yield at 900 °C (31.0%) which is due to the lower C content and higher carbohydrates content and the lower stability of carbohydrates at 900 °C. All lignins obtained after 90 min had a yield higher than 36%. The higher yield at 900 °C was in agreement with the elemental analysis (Table 1) and confirmed that lignins fractionated at longer times contained a higher C content. In case of using lignin as carbon precursor of carbon fiber or active carbon, the lignin with higher char yield has the economic and environmental advantages.40 The main decomposition temperature increased from 369 °C (30 min) to 384 °C (120 min) with fractionation time also indicating improved purity with a decrease in polysaccharide content. An intermediate decomposition temperature was observed for the control (376 °C), which is normally the mean for all lignin samples. When lignin is used as a carbon precursor, thermal pretreatment is usually applied at 150 to 270 °C to enable the fiber formation.43 Therefore, lignins with higher decomposition temperature are desired. The higher thermal stability found in the lignins fractionated at later time indicated that those fractions with enhanced thermal stability would be capable of various applications.
Pearson correlation test
In order to reveal relationships between the measured characteristics of the fractionated lignins, a Pearson correlation test was performed (ESI Table S1†). The associated p-value is an indicator of whether a given correlation coefficient is statistically significant. In this study, all the tests were performed at significant level of 0.05. A p-value <0.05 confirms a significant linear correlation between two variables. The Pearson correlation test indicates that the chemical characteristics of the lignin samples were strongly correlated to the thermal properties, such as Tg. Fig. 4 shows some of the major significant correlations identified by the Pearson test. Lignin content was positively correlated to Tg whereas hemicellulose content was negatively correlated. Positive correlations were also found between total phenolic OH and Tg as well as syringyl OH concentration and Tg, while negative correlation was found between aliphatic OH concentration and Tg (Fig. 4). These results indicated the formation of hydrogen bonds through a total increase in OH content and cleavage of ether bond might lead to higher Tg of lignin. No real measure of condensed carbon structure was performed; however, the correlation between ether bonds cleavage with increased Tg could be indicative of increased crosslinking with time. From the Pearson correlation table, the Mw and Mn strongly correlated with the purity of the lignin, but did not have any statistically significant correlation with the thermal properties measured. Since the PDI of the lignin ranges from 3.6 to 6.6, the broad distribution of the lignin molecular weight might cause the invalidity in the correlation calculation. In summary, the Pearson correlation revealed that purity and hydroxyl content had the greatest effect on lignin thermal properties.
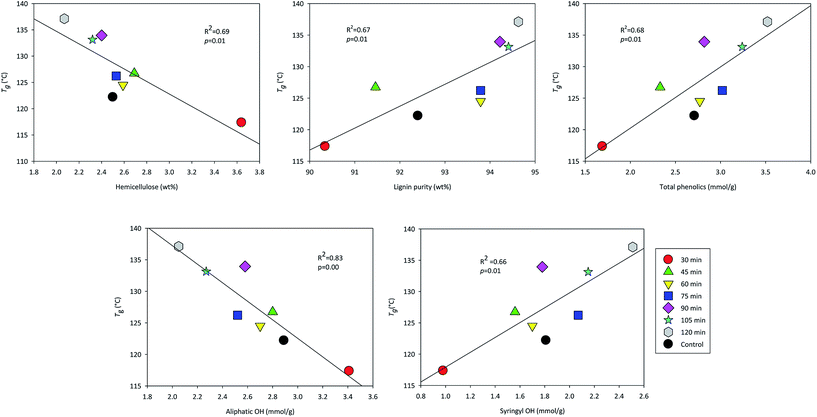 |
| | Fig. 4 The correlation between lignin chemical properties (hemicellulose content, lignin content, total phenolic OH, aliphatic OH, and syringyl OH) and Tg. | |
Conclusions
The analysis of lignins generated by fractionation at different times during an organosolv process showed that about 70% of the lignin present in the biomass can be collected within 90 min. This result suggests that a fractionation time of 90 min could maintain a relatively high level of lignin yield while reducing solvent consumption by 20% and lowering energy usage. More importantly, lignin purity dramatically improves after the first 30 min. At longer fractionation times, phenolic and total hydroxyl content of lignin increased, while ether linkages decreased leading to more condensed lignin structure with higher Tg's. Overall, the molecular weight tended to decrease and become broader at long times as the PDI increased. This progressive shift in lignin chemical structure to a more condensed phenolic form with time during organosolv will have important implications on the end use and functionality of the resulting lignin. The structures formed at longer fractionation times (>90 min) will likely be less fusible, reactive, and therefore less valuable for further chemical transformation.
Acknowledgements
This project is funded by the University of Tennessee Institute of Agriculture and the Southeastern Partnership for Integrated Biomass Supply Systems supported by the Agriculture and Food Research Initiative (Competitive Grant number 2011-68005-30410).
References
- S. K. Black, B. R. Hames, M. D. Myers, Method of separating lignocellulosic material into lignin, cellulose and dissolved sugars, US Pat., US 5730837, 1998 Mar 24.
- E. K. Pye and J. Lora, Tappi J., 1991, 74, 113–118 CAS.
- S. Constant, H. L. J. Wienk, A. E. Frissen, P. D. Peinder, R. Boelens, D. S. Van Es, R. J. H. Grisel, B. M. Weckhuysen, W. J. J. Huijgen, R. J. A. Gosselink and P. C. A. Bruijnincx, Green Chem., 2016, 18, 2651–2665 RSC.
- J. J. Bozell, S. K. Black, M. Myers, D. Cahill, W. P. Miller and S. Park, Biomass Bioenergy, 2011, 35, 4197–4208 CrossRef CAS.
- S. Chatterjee, T. Saito, O. Rios and A. Johs, in Green Technologies for the Environment, American Chemical Society, 2014, vol. 1186, ch. 11, pp. 203–218 Search PubMed.
- D. A. Baker and T. G. Rials, J. Appl. Polym. Sci., 2013, 130, 713–728 CrossRef CAS.
- S. Kubo and J. F. Kadla, Macromolecules, 2004, 37, 6904–6911 CrossRef CAS.
- R. J. Sammons, D. P. Harper, N. Labbé, J. J. Bozell, T. Elder and T. G. Rials, BioResources, 2013, 8, 2752–2767 CrossRef.
- W. J. J. Huijgen, G. Telysheva, A. Arshanitsa, R. J. A. Gosselink and P. J. de Wild, Ind. Crops Prod., 2014, 59, 85–95 CrossRef CAS.
- X. Pan, J. F. Kadla, K. Ehara, N. Gilkes and J. N. Saddler, J. Agric. Food Chem., 2006, 54, 5806–5813 CrossRef CAS PubMed.
- F. Xu, J. X. Sun, R. Sun, P. Fowler and M. S. Baird, Ind. Crops Prod., 2006, 23, 180–193 CrossRef CAS.
- T. Shui, S. Feng, Z. Yuan, T. Kuboki and C. Xu, Bioresour. Technol., 2016, 218, 953–961 CrossRef CAS PubMed.
- J. Snelders, E. Dornez, B. Benjelloun-Mlayah, W. J. J. Huijgen, P. J. de Wild, R. J. A. Gosselink, J. Gerritsma and C. M. Courtin, Bioresour. Technol., 2014, 156, 275–282 CrossRef CAS PubMed.
- L. Jiménez, M. J. De La Torre, J. L. Bonilla and J. L. Ferrer, Process Biochem., 1998, 33, 401–408 CrossRef.
- Y. Jafari, H. Amiri and K. Karimi, Appl. Energy, 2016, 168, 216–225 CrossRef CAS.
- W. J. J. Huijgen, J. H. Reith and H. den Uil, Ind. Eng. Chem. Res., 2010, 49, 10132–10140 CrossRef CAS.
- A. Rodríguez, L. Serrano, A. Moral and L. Jiménez, Biochem. Eng. J., 2008, 42, 243–247 CrossRef.
- H. Kangas, T. Liitiä, S. Rovio, T. Ohra-Aho, H. Heikkinen, T. Tamminen and K. Poppius-Levlin, Holzforschung, 2015, 69, 247–256 CrossRef CAS.
- G. Gellerstedt and E.-L. Lindfors, International Journal of the Biology, Chemistry, Physics, and Technology of Wood, 1984, 38, 151–158 CrossRef CAS.
- R. El Hage, N. Brosse, P. Sannigrahi and A. Ragauskas, Polym. Degrad. Stab., 2010, 95, 997–1003 CrossRef CAS.
- J. J. Bozell, C. J. O'Lenick and S. Warwick, J. Agric. Food Chem., 2011, 59, 9232–9242 CrossRef CAS PubMed.
- S. Y. Lin and C. W. Dence, Methods in Lignin Chemistry, Springer, Berlin Heidelberg, 2011 Search PubMed.
- A. Granata and D. S. Argyropoulos, J. Agric. Food Chem., 1995, 43, 1538–1544 CrossRef CAS.
- Y. Pu, S. Cao and A. J. Ragauskas, Energy Environ. Sci., 2011, 4, 3154–3166 CAS.
- G. Hu, C. Cateto, Y. Pu, R. Samuel and A. J. Ragauskas, Energy Fuels, 2012, 26, 740–745 CrossRef CAS.
- P. Liu, L. Yu, H. Liu, L. Chen and L. Li, Carbohydr. Polym., 2009, 77, 250–253 CrossRef CAS.
- X. Pan, N. Gilkes, J. Kadla, K. Pye, S. Saka, D. Gregg, K. Ehara, D. Xie, D. Lam and J. Saddler, Biotechnol. Bioeng., 2006, 94, 851–861 CrossRef CAS PubMed.
- H. L. Chum, D. K. Johnson and S. K. Black, Ind. Eng. Chem. Res., 1990, 29, 156–162 CrossRef CAS.
- J. M. Rosas, R. Berenguer, M. J. Valero-Romero, J. Rodriguez-Mirasol and T. Cordero, Front. Mater., 2014, 1(29) DOI:10.3389/fmats.2014.00029.
- Q. Wang, S. Liu, G. Yang and J. Chen, Energy Fuels, 2014, 28, 3167–3171 CrossRef CAS.
- T. Leskinen, S. S. Kelley and D. S. Argyropoulos, ACS Sustainable Chem. Eng., 2015, 3, 1632–1641 CrossRef CAS.
- J. Gierer, Wood Sci. Technol., 1985, 19, 289–312 CAS.
- R. El Hage, N. Brosse, L. Chrusciel, C. Sanchez, P. Sannigrahi and A. Ragauskas, Polym. Degrad. Stab., 2009, 94, 1632–1638 CrossRef CAS.
- F. Monteil-Rivera, M. Phuong, M. Ye, A. Halasz and J. Hawari, Ind. Crops Prod., 2013, 41, 356–364 CrossRef CAS.
- X. Pan and J. N. Saddler, Biotechnol. Biofuels, 2013, 12 CrossRef CAS PubMed.
- C. Pouteau, P. Dole, B. Cathala, L. Averous and N. Boquillon, Polym. Degrad. Stab., 2003, 81, 9–18 CrossRef CAS.
- O. Faix, International Journal of the Biology, Chemistry, Physics, and Technology of Wood, 1991, 45, 21–28 CAS.
- C. Heitner, D. Dimmel and J. Schmidt, Lignin and Lignans: Advances in Chemistry, Taylor & Francis, 2011 Search PubMed.
- L. M. Kline, D. G. Hayes, A. R. Womac and N. Labbé, BioResources, 2010, 5, 1366–1383 CAS.
- T. Saito, J. H. Perkins, F. Vautard, H. M. Meyer, J. M. Messman, B. Tolnai and A. K. Naskar, ChemSusChem, 2014, 7, 221–228 CrossRef CAS PubMed.
- H.-W. Kammer, Physical Chemistry of Macromolecules: Macro to Nanoscales, Apple Academic Press, 2014 Search PubMed.
- Y. N. Sazanov and A. Gribanov, Russ. J. Appl. Chem., 2010, 83, 175–194 CrossRef CAS.
- J. F. Kadla, S. Kubo, R. A. Venditti, R. D. Gilbert, A. L. Compere and W. Griffith, Carbon, 2002, 40, 2913–2920 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra16296g |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.