
Selectfluor-mediated highly selective radical dioxygenation of alkenes†
Gang
Li,  a
a
a
Abstract
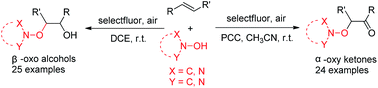
A practical and simple selectfluor mediated highly selective radical dioxygenation of alkenes was achieved under mild conditions. Various hydroxylamines, such as N-hydroxyphthalimide (NHPI), N-hydroxybenzotriazole (HOBt) and N-hydroxysuccinimide (NHSI), could react smoothly with alkenes to give β-oxo alcohols and α-oxy ketones in good to moderate yields. The reaction mechanism was primarily investigated and a radical process was proposed.
 Please wait while we load your content...
Please wait while we load your content...

