
A practical and efficient route to heteraphanes: synthesis of structurally simplified analogues of ansamycins†
R. Santhosh Reddy‡
,
Shaojun Zheng‡,
Chandraiah Lagishetti,
Hengyao You and
Yun He*
School of Pharmaceutical Sciences and Innovative Drug Research Centre, Chongqing University, 55 Daxuecheng South Rd., Shapingba, Chongqing 401331, P.R. China. E-mail: yun.he@cqu.edu.cn
First published on 6th July 2016
Abstract
A highly efficient and versatile synthetic strategy has been developed for the synthesis of meta-, para-azabenzeno or azanaphthaleno phanes (II–IV) with varying ring sizes (12–25) through intramolecular Mitsunobu reaction from the corresponding Ns-arylamino alcohols. The reaction is compatible with olefin and ketone moieties in the backbone tether and has been applied in the synthesis of various simplified structural motifs of ansamycins.
The ansamycins comprise a diverse group of complex, often remarkably bioactive and historically important natural products that have been known for over 50 years.1 A hallmark of these compounds is a medium- to large-sized macrocycle “handle” fused to a mono- or bicyclic aromatic core. Based on the aromatic chromophore and the length of the ansa chain (the ring size of the macrocycle), most of the important ansamycins have been classified as shown in Fig. 1a.2 Among the most prominent representatives with complex chemical architectures and potent biological activities are antimycobacterial antibiotic rifamycin,3 polypeptide biosynthesis inhibitor rubradirin,4 Hsp90 inhibitor geldanamycin,5 and antiproliferative maytansinoids,6 etc.7,8 In addition to the antibacterial, antitumor and herbicidal properties that many ansamycins exhibit, azanaphthaleno or azabenzenophane (heteraphane)9 structural motifs (see II and III, Fig. 1c) may be of interest to chemical biology and medicinal chemistry as novel molecular diversity whose biological properties remain largely unexplored.10 For example, rubradirins (1a–c), neoansamycins (2a–b), and geldanamycin (3) are such molecules and their origins can be possibly traced back to azanaphthaleno or azabanzeno phanes 4, 5 and 6 respectively as shown in Fig. 1b.11 Therefore, strategies for the efficient synthesis of these fused macrocycles are crucial to the total synthesis of the natural products, chemical and medicinal studies of natural and designed ansamycins.
 | ||
| Fig. 1 (a) Classification of ansamycins; (b) structures of ansamycins (1–3) and their hypothetical synthetic precursors (4–6) (c) structures of heteraphane (I) and targeted aza naphthalenophane, m-, and p-azabenzenophanes (II, III and IV). | ||
As part of our research program directed towards the synthesis of complex ansamycins, the formation of the 15-membered azanaphthaleno phane (II) as a model system for rubradirin synthesis was examined using common cyclization methods such as ring-closing metathesis, macrolactamization, cross-couplings, Wittig olefination, reductive amination and alkylation. Unfortunately, all these efforts failed to provide the desired macrocycle.12 Fukuyama and co-workers have applied intramolecular Mitsunobu reaction to prepare an 11-memebered cyclic intermediate in the synthesis of (+)-vinblastine.13b To our delight, the intramolecular Mitsunobu reaction13 using TMAD and nBu3P was found to be highly efficient for the synthesis of the desired 15-membered azanaphthaleno phane (II). We thus set out to conduct systematic studies on its scope and potential in synthesizing various natural and designed ansamycins. Herein we report a practical, versatile, selective and general strategy for the construction of azanaphthalenophanes (II), m-azabenzenophanes (III) and p-azabenzenophanes (IV) via intramolecular Mitsunobu reaction of the corresponding Ns-arylamino alcohol derivatives.
Table 1 shows our studies to optimize macrocyclisation conditions for the formation of the 15-membered aza naphthalenophane 8 from the corresponding substrate Ns-arylamino alcohol 7. In the initial explorations, TMAD13c (1.5 equiv.) and nBu3P (1.5 equiv.) were added to a solution of substrate 7 in toluene (0.05 M, entry 1) at 80 °C, and the corresponding macrocyclic product 8 was isolated in 48% yield as the sole product. Under higher dilution conditions (i.e. 0.025 M and 0.005 M, entries 2 and 3), the reaction led to improved yields, giving rise to the desired macrocycle 8 in 55% and 78% yield respectively. Macrocyclization didn‘t occur at room temperature in toluene or THF (entries 4 and 5). Increasing the amount of TMAD and nBu3P (3 equiv. and 6 equiv., entries 6 & 7) reduced the reaction time but didn‘t improve the yield. A brief evaluation of solvents (THF, benzene and toluene) suggested that toluene was the most suitable one. When DIAD or DEAD was used in the place of TMAD, neither reaction produced the desired macrocycle (entries 8 and 9). Efforts to establish a catalytic process were also attempted,14 when the reaction was carried out with an oxidant [TMAD (0.5 equiv.), nBu3P (0.5 equiv.), PhI(OAc)2 (2.0 equiv.)] in toluene, the desired product 8 was obtained in 10% yield (entry 10). Thus entry 3 represented the optimized condition, which was used for further studies.
|
|||||
|---|---|---|---|---|---|
| Entry | Reagentb (equiv.) | Solvent | Conc. (M) | T (°C)/t (h) | Yieldc (%) |
| a Reactions were performed on 50 mg scale.b Reagents: A: TMAD, nBu3P; B: DIAD, PPh3; C: DEAD, PPh3.c Isolated yield after column chromatographic purification.d No reaction observed.e Not detected by TLC.f Oxidant PhI(OAc)2 (2.0 equiv.) was used. | |||||
| 1 | A (1.5) | Toluene | 0.05 | 80/6 | 48 |
| 2 | A (1.5) | Toluene | 0.025 | 80/6 | 55 |
| 3 | A (1.5) | Toluene | 0.005 | 80/8 | 78 |
| 4d | A (1.5) | Toluene | 0.005 | 25/12 | n.r. |
| 5d | A (1.5) | THF | 0.005 | 25/12 | n.r. |
| 6 | A (3.0) | Toluene | 0.005 | 80/4 | 68 |
| 7 | A (6.0) | Toluene | 0.005 | 80/2 | 69 |
| 8d | B (1.5) | THF | 0.005 | 25/12 | n.r. |
| 9e | C (1.5) | Toluene | 0.005 | 80/12 | n.d. |
| 10f | A (0.5) | Toluene | 0.05 | 80/12 | 10 |

In order to explore the generality and scope of this macrocyclization reaction, a range of substrates were prepared and subjected to the optimized conditions, leading to a variety of macrocyclic systems containing the aza naphthalenophane structural motif. Table 2 summarizes the results of these investigations. It was not surprising, but of interest that the formation of 14-membered ring aza naphthalenophane (9) was not observed from the corresponding Ns-arylamino alcohol, even under the high dilution conditions, presumably due to the severe transannular interactions and bond angle distortions which develops in the transition state for ring closure in the medium-sized ring system.15 Larger rings (i.e. 16-, 17- and 21-membered) were formed in good yields as expected (10, 11 and 12, 68%, 69% and 71% respectively). The introduction of trans olefinic bond in the aliphatic tether of the precursor had no significant effect on the cyclization, leading to the 15-membered macrocycle 13 in 70% yield. Similarly, the introduction of trans olefinic bond within the long chain aliphatic linker underwent smooth macrocyclization to provide 18-, 20- and 23-membered heteraphanes in higher yields (14, 15 and 16, 74%, 74% and 76%, respectively). It is noteworthy to mention that, the obtained 15-, 17-, and 23-membered azanaphthalenophanes are the simplified structural motifs of bioactive rubradirin, neoansamycin and protorifamycin, etc.8 respectively.
|
|---|
| a Reactions were performed on ca. 100 mg scale as follows: TMAD (1.5 equiv.), nBu3P (1.5 equiv.) was added to a substrate (1.0 equiv., 0.005 M) in toluene at RT and the reaction then keep at 80 °C for 8 h.b Isolated yield after column chromatographic purification.c For the preparation of substrates and further details, see ESI. |
 |
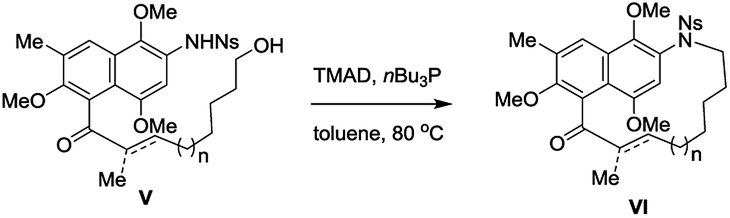
Encouraged by these results, we proceeded to examine the formation of various m-azabenzenophanes structural motifs which are featured in the benzenoid ansamycins. Table 3 shows the results of intramolecular macrocyclization of Ns-amino alcohols VII. The ring closure of these substrates was anticipated to be easier than the azanaphthalenophanes (see Table 2) due to a single aromatic ring within the carbon frame work. Not surprisingly, the formation of 11-membered m-azabenzenophane (17) failed from the corresponding Ns-arylamino alcohol under the optimized reaction condition, presumably due to the severe strain and transannular interactions associated with this medium-sized rings.15 Other medium-sized rings (12-, 13-, and 14-membered) notable for their intransigence of formation (18, 19 and 20) were generated, in moderate to good yields as shown in Table 3. Larger rings (15-, 16-, 19- and 24-membered) were formed in higher yields, as expected (21, 22, 23 and 24; 78%, 76%, 78% and 84%, respectively). The aliphatic linker with α,β-unsaturated ketone proceeded efficiently to furnish the 21-membered m-azabenzenophane 25 in high yield. The obtained 19-, and 21-membered azabenzenophanes are the simplified structural analogues of bioactive natural products ansamitocin, geldanamycin, etc., and ansatrienin, cytotrienin, etc.7 respectively.

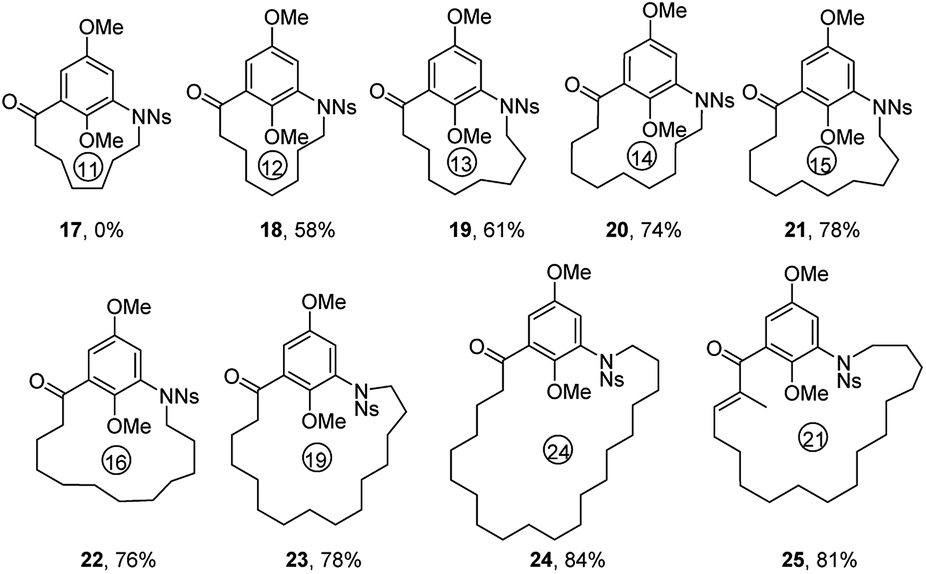
Subsequently, we extended our studies to include other Ns-arylamino alcohol derivatives (IX) bearing the ansa chains at para position of Ns-arylamine under the optimized reaction condition to generate novel molecular diversity (Table 4). Similar to the formation of 17, the 13-membered p-azabenzenophane ring (26) was not observed under the optimized reaction condition. On the other hand, under the same reaction condition, macrocycles 27–31 (14-, 15-, 16-, 17-, 20-membered) were formed in good to excellent yields. Substrate with a 20-carbon ansa chain also furnished the corresponding benzenophane 32 in 85% yield.

To further demonstrate the synthetic utility of this synthetic technology, model synthesis of the core macrocyclic skeleton of rubradirins (1a–c) was carried out.4 The first member of the rubradirin family, rubradirin (1a), an ansamycin was isolated in 1964 and the other two congeners, rubradirins B and C (1b & 1c) were isolated in 1978 and 1982 respectively.16 Despite the presence of complex and unique molecular architectures associated with interesting biological activity profiles, surprisingly, no total synthesis of any member of rubradirin family has been reported thus far. Herein we report the construction of a hexa substituted naphthalene derivative containing an aliphatic ansa bridge bereft of chiral functionality but of the same ring size in rubradirins.17 As shown in Scheme 1, the organolithium reagent generated from naphthalenic core 33 was obtained by halogen-metal exchange using nBuLi (−100 °C, 20 min) and was then reacted with α,β-unsaturated aldehyde 34 (THF, −100 °C, 1.5 h) to produce the alcohol with good yield, which upon oxidation under DMP conditions provided the corresponding ketone in 78% yield. Subsequent deprotection of TBS group was accomplished in excellent yield by TBAF. The resulting alcohol was in turn converted to aldehyde 35 in 91% yield.18 Aldehyde 35 was reacted with the stabilized Wittig reagent to produce the (E)-α,β-unsaturated ester in 88% yield, and further Boc deprotection was carried out with TFA to furnish the corresponding amine in 81% yield. The obtained free amine was first protected with NsCl and the ethyl ester was then reduced with DIBALH in THF to alcohol 7 in 59% yield along with saturated ketone 36 in 24% yield. With the key intermediates 7 and 36 in hand, the synthesis of the azanaphthaleno phanes was examined. Under the optimized condition, the intramolecular Mitsunobu macrocyclization of Ns-amino alcohols 7 and 36 provided the 15-membered aza naphthalenophanes 8 and 37 in 78% and 76% yield respectively. Aza naphthalenophanes 8 and 37 possess the complete carbon frame work present in rubradirin macrocycles. Oxidation of the dimethoxy aromatic core in 8 with CAN furnished the corresponding quinone, and deprotection of nosyl group19 resulted in the formation of amino quinone 38 in 72% yield. To test the practicality of our synthetic strategy, a gram scale synthesis of aza naphthalenophane 8 was carried out. To our delight, the 2.6 mmol scale reaction produced 1.21 grams of 8 in 76% yield, suggesting that this transformation is amenable to scale up without loss of efficiency. Aza naphthalenophane 8 could be crystalized from CH2Cl2/MeOH (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2) and its structure was confirmed by X-ray crystallographic analysis.20
:2) and its structure was confirmed by X-ray crystallographic analysis.20
 | ||
Scheme 1 Application of the Mitsunobu strategy in the model synthesis of rubradirin macrocycle. Reagents and conditions: (a) nBuLi, 33, THF, −100 °C, 20 min; then 34, −100 °C to −78 °C, 1.5 h, 72%; (b) DMP, CH2Cl2, 25 °C, 15 min, 78%; (c) TBAF, THF, 25 °C, 12 h, 82%; (d) IBX, EtOAc, reflux, 6 h, 91%; (e) Ph3P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(Me)CO2Et, PhCH3, reflux, 8 h, 88%; (f) TFA:CH2Cl2 (1:3), 25 °C, 10 h, 81%; (g) NsCl, py, 25 °C, 8 h, 92%; (h) DIBALH, THF, −78 °C, 12 h, 59% (7), 24% (36); (i) TMAD, nBu3P, PhCH3, 80 °C, 8 h 78% for 8 and 76% for 37; (j) CAN, CH3CN:H2O (9:1), 25 °C, 12 h; 45%; (k) CH3PhSH, K2CO3, DMF, 0 °C, 1 h, 72%. C(Me)CO2Et, PhCH3, reflux, 8 h, 88%; (f) TFA:CH2Cl2 (1:3), 25 °C, 10 h, 81%; (g) NsCl, py, 25 °C, 8 h, 92%; (h) DIBALH, THF, −78 °C, 12 h, 59% (7), 24% (36); (i) TMAD, nBu3P, PhCH3, 80 °C, 8 h 78% for 8 and 76% for 37; (j) CAN, CH3CN:H2O (9:1), 25 °C, 12 h; 45%; (k) CH3PhSH, K2CO3, DMF, 0 °C, 1 h, 72%. | ||
In summary, a versatile, efficient and general strategy for the practical synthesis of a wide variety of heteraphanes through intramolecular Mitsunobu reaction from readily available simple Ns-arylamino alcohols has been developed. Products obtained through this novel cyclization include macrocyclic quinone intermediates relevant to biosynthetic hypothesis21 and total synthesis of the ansamycin family of natural products. This strategy enables our efforts towards the synthesis of natural, designed rubradirins12 and other bioactive ansamycin molecules.11 It is also expected to find further applications as a useful technology in chemical biology and medicinal chemistry investigations. These efforts are on-going in our laboratories and shall be reported in due course.
Acknowledgements
We are grateful for the financial support from the National Natural Science Foundation of China (No. 21572027, and No. 21372267) and the Postdoctoral Research Grant (2014M562278 and Xm2014002).Notes and references
- A. Bryskier, Antimicrobial Agents: Antibacterials and Antifungals, ASM Press, Washington DC, 2005 Search PubMed.
- S. Funayama and G. A. Cordell, Studies in Natural Products Chemistry, ed. Atta-ur-Rahman, Elsevier Science Publishers, Amsterdam B. V, 2000, vol. 23, pp. 51–106 Search PubMed.
- H. G. Floss and T. W. Yu, Chem. Rev., 2005, 105, 621 CrossRef CAS PubMed.
- (a) C. E. Meyer, Antimicrob. Agents Chemother., 1964, 97 CAS; (b) F. Reusser, Biochemistry, 1973, 12, 1136 CrossRef CAS PubMed; (c) F. Reusser, J. Antibiot., 1979, 32, 1186 CrossRef CAS PubMed.
- L. Whitesell, E. G. Mimnaugh, B. De Costa, C. E. Myers and L. M. Neckers, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 8324 CrossRef CAS.
- J. M. Cassady, K. K. Chan, H. G. Floss and E. Leistner, Chem. Pharm. Bull., 2004, 52, 1 CrossRef CAS PubMed.
- For recent reviews on the biological activity of benzoquinone ansamycins, see: (a) W. Xu and L. Neckers, Clin. Cancer Res., 2007, 13, 1625 CrossRef CAS PubMed; (b) Y. Miyata, Curr. Pharm. Des., 2005, 11, 1131 CrossRef CAS PubMed.
- For isolation and biological activity of naphthoquinone ansamycins, see: (a) S. Matsuda, K. Adachi, Y. Matsuo, M. Nukina and Y. Shizuri, J. Antibiot., 2009, 62, 519 CrossRef CAS PubMed; (b) P. Cai, F. Kong, M. E. Ruppen, G. Glasier and G. T. Carter, J. Nat. Prod., 2005, 68, 1736 CrossRef CAS PubMed; (c) C. Lu and Y. Shen, J. Antibiot., 2007, 60, 649 CrossRef CAS PubMed; (d) M. C. Wilson, S.-J. Nam, T. A. M. Gulder, C. A. Kauffman, P. R. Jensen, W. Fenical and B. S. Moore, J. Am. Chem. Soc., 2011, 133, 1971 CrossRef CAS PubMed; (e) L. Ding, A. Maier, H.-H. Fiebig, H. Grls, W.-H. Lin, G. Peschel and C. Hertweck, Angew. Chem., Int. Ed., 2011, 50, 1630 CrossRef CAS PubMed; (f) S. Li, Y. Li, C. Lu, J. Zhang, J. Zhu, H. Wang and Y. Shen, Org. Lett., 2015, 17, 3706 CrossRef CAS PubMed.
- R. Gleiter and H. Hopf, Modern Cyclophane Chemistry, Wiley-VCH, Weinheim, 2004, vol. 45, pp. 41–73, 1–5 Search PubMed.
- M. M. Madathil, O. M. Khdour, J. Jaruvangsanti and S. M. Hecht, ACS Med. Chem. Lett., 2013, 4, 953 CrossRef CAS PubMed.
- Conversion of amine to amide see: (a) K.-I. Tanaka, S. Yoshifuji and Y. Nitta, Chem. Pharm. Bull., 1988, 36, 3125 CrossRef CAS; (b) Y. Wang, H. Kobayashi, K. Yamaguchi and N. Mizuno, Chem. Commun., 2012, 48, 2642 RSC; (c) J. R. Khusnutdinova, Y. Ben-David and D. Milstein, J. Am. Chem. Soc., 2014, 136, 2998 CrossRef CAS PubMed.
- Unpublished results, this laboratory.
- (a) K. C. K. Swamy, N. N. B. Kumar, E. Balaraman and K. V. P. P. Kumar, Chem. Rev., 2009, 109, 2551 CrossRef CAS PubMed; (b) T. Miyazaki, S. Yokoshima, S. Simizu, H. Osada, H. Tokuyama and T. Fukuyama, Org. Lett., 2007, 9, 4737 CrossRef CAS PubMed; (c) T. Tsunoda, J. Otsuka, Y. Yamamiya and S. Ito, Chem. Lett., 1994, 539 CrossRef CAS; (d) T. Kan and T. Fukuyama, Chem. Commun., 2004, 353 RSC.
- T. Y. S. But and P. H. Toy, J. Am. Chem. Soc., 2006, 128, 9636 CrossRef CAS PubMed.
- P. Deslongchamps, S. Lamothe and H.-S. Lin, Can. J. Chem., 1984, 62, 2395 CrossRef CAS.
- (a) H. Hoeksema, C. Lewis, S. A. Mizsak, J. A. Shiley, D. R. Wait, H. A. Whaley and G. E. Zurenko, J. Antibiot., 1978, 31, 945 CrossRef CAS PubMed; (b) H. Hoeksema, S. A. Mizsak, L. Baczynskyj and L. M. Pschigoda, J. Am. Chem. Soc., 1982, 104, 5173 CrossRef CAS.
- A. P. Kozikowski and Y. Xia, J. Org. Chem., 1987, 52, 1375 CrossRef CAS.
- M. Frigerio, M. Santagostino and S. Sputore, J. Org. Chem., 1999, 64, 4537 CrossRef CAS.
- T. Kan and T. Fukuyama, Chem. Commun., 2004, 353 RSC.
- CCDC number for 8: 1456426.†.
- Q. Kang, Y. Shen and L. Bai, Nat. Prod. Rep., 2012, 29, 243 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, full characterization, and copies of NMR spectra of new compounds. CCDC 1456426. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra16247a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2016 |